Clear Sky Science · sv
Dubbel maskininlärning identifierar radien för informativa strukturella miljöer i metalliska glas
Varför denna dolda skala i glas spelar roll
Metalliska glas är metaller som kylts så snabbt att de aldrig bildar en kristall. Denna avsaknad av regelbunden struktur ger dem ovanlig styrka och seghet, men gör dem också svåra att avsiktligt utforma: utan ett prydligt kristallmönster har det varit oklart vilka delar av deras atomära struktur som verkligen styr deras beteende. Denna artikel använder avancerad maskininlärning för att visa att det faktiskt finns en optimal storlek i dessa oordnade material — en slags "Guldlock"-närmiljö runt varje atom — där strukturen innehåller den mest användbara informationen om hur hela materialet kommer att bete sig.
Att se ordning i oordnade metaller
I vanliga kristallina metaller kan ingenjörer koppla styrka och duktilitet till välkända egenskaper som kornstorlek eller dislokationer, vilka sträcker sig över många atomer i ett regelbundet gitter. Metalliska glas saknar den långväga ordningen och de uppenbara defekterna, så forskare har försökt förlita sig på mer lokala byggstenar kallade kortväga ordningar — hur atomer packar runt en central atom. Tidigare arbete visade att dessa närmaste grannar inte räcker: samma lokala motiv kan bete sig mycket olika beroende på omgivningen. Det pekade på en avgörande öppen fråga: över vilken räckvidd runt varje atom behöver vi titta för att fånga de strukturella mönster som faktiskt kontrollerar makroskopiska egenskaper som styrka och stabilitet?
En top-down blick på atomära närmiljöer
Författarna börjar med ett reduktionistiskt, eller top-down, angreppssätt. De representerar varje atoms omgivning som ett matematiskt fingeravtryck och grupperar liknande fingeravtryck i en uppsättning unika lokala miljöer. För varje prov av metalliskt glas räknar de hur ofta varje miljö förekommer och tränar en maskininlärningsmodell (XGBoost) för att förutsäga provets totala konfigurationsenergi, en storhet nära kopplad till hur starkt eller duktilt materialet är. Viktigt är att de upprepar hela denna process samtidigt som de varierar hur långt från varje atom de tittar — från bara de första grannarna till avstånd som omfattar flera grannskal. Modellens prediktionsfel förbättras inte helt enkelt när fler atomer inkluderas. Istället når den sin bästa prestanda när radien är omkring 5 ångström, ungefär ut till andra grannskalet, och försämras därefter igen. Vid den radien når antalet och mångfalden av distinkta lokala miljöer en topp, och spridningen i en strukturell entropimått är som störst, vilket indikerar att denna skala packar in den rikaste strukturella informationen.
En bottom-up vy från 3D-bilder
För att testa om denna speciella radie kan vara en artefakt av deras första metod bygger teamet en andra, mycket annorlunda modell inspirerad av modern bildigenkänning. De omvandlar varje atomstruktur till ett par tredimensionella pixlarnät — en kanal för varje kemisk art — och matar dessa till en vision transformer, ett neuralt nätverk som lär sig mönster i rutblock av nätet. Genom att ställa in hur långt dessa patchar får "prata" med varandra styr de en effektiv kommunikationslängd som sätter den största strukturella skala modellen kan använda. När de ökar denna längd förbättras modellens noggrannhet men planar sedan ut: bortom en kommunikationslängd motsvarande en sfärisk radie på cirka 5 ångström ger ytterligare räckvidd nästan ingen nytta. Oberoende analys av vilka regioner i nätet som mest påverkar prediktionen visar att nätverkets attention också konvergerar när denna samma skala uppnås.
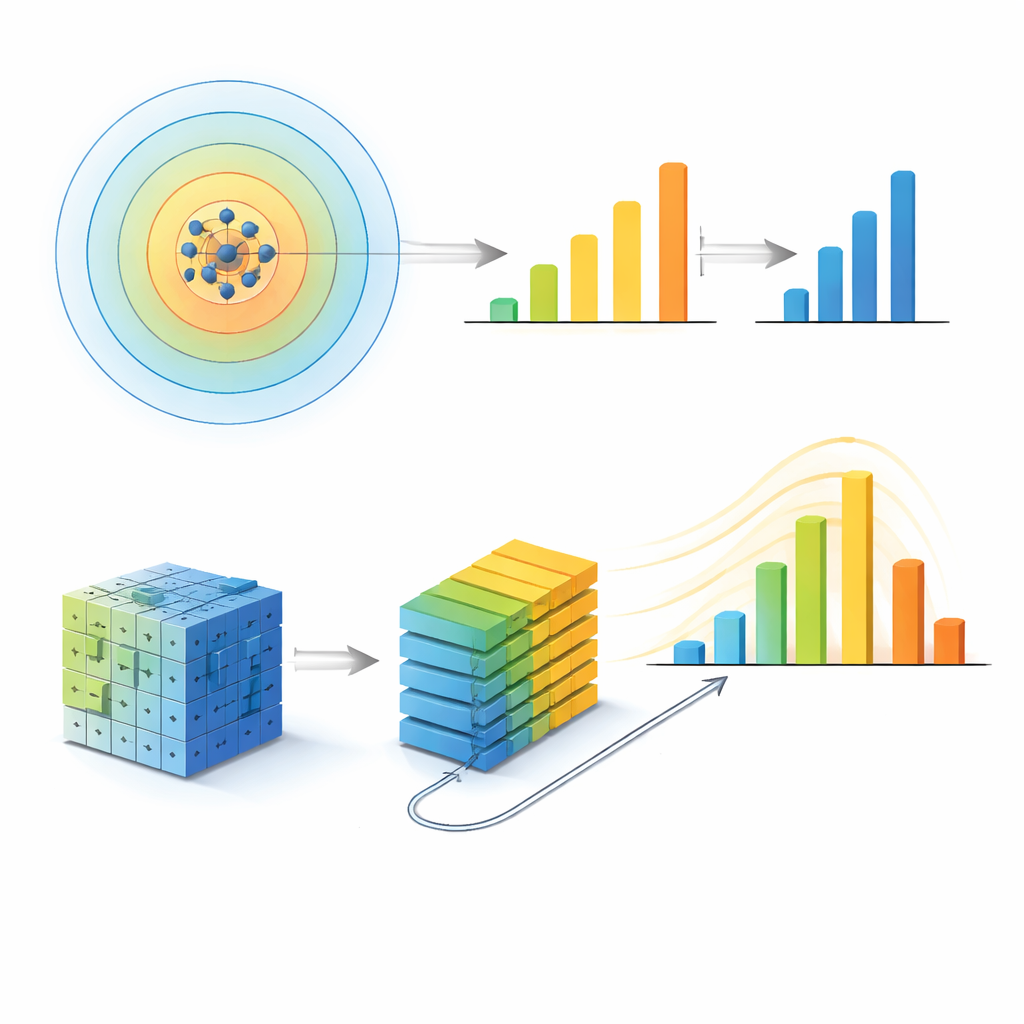
Robust över storlekar och olika glas
Överensstämmelsen mellan dessa två kontrasterande maskininlärningsvyer — den ena byggd på handgjorda atomära fingeravtryck, den andra på råa vokselgaller — tyder på att den identifierade längdskalan är en verklig fysisk egenskap, inte en modelleringskuriosa. Författarna utsätter vidare denna idé för stressprov genom att ändra tekniska inställningar, systemstorlekar och till och med typerna av glasmaterial. Större simulationsboxar med fler atomer bevarar samma optimala radie. När de upprepar analysen för mer komplexa metalliska glas som innehåller aluminium, och för ett kemiskt skilt palladium–kisel-glas, visar varje system återigen en specifik informativ radie, som är något mindre i det tätare packade Pd–Si-fallet. Liknande beteende framträder i amorft kol och kisel, där kovalenta bindningar leder till olika men fortfarande väl definierade karaktäristiska skalor. I samtliga dessa fall linjerar den radie där modellprestationen når topp eller mättnad med oberoende strukturella och experimentella ledtrådar om medelräckviddsorganisation i dessa material.
Vad studien avslöjar i enkla termer
För en icke-specialist är huvudbudskapet att även i ett till synes slumpmässigt metalliskt glas finns en naturlig "inflytandeszon" runt varje atom — ungefär två atomer ut — där ordningen hos grannarna bär nästan all information som behövs för att förutsäga hur hela glasstycket kommer att bete sig energimässigt. Att endast betrakta de allra närmaste grannarna missar viktig kontext, medan att titta för långt ut bara lägger till redundant detalj. Genom att peka ut denna radie för informativa strukturella miljöer ger arbetet en praktisk målskala för framtida modeller och experiment. Det erbjuder en vägkarta för att utforma bättre metalliska glas och andra amorfa material genom att fokusera uppmärksamheten på de strukturella motiv som lever på denna mellanliggande skala, där dold ordning i oordning starkast formar makroskopiska egenskaper.
Citering: Wang, M., Wang, Y., Islam, M. et al. Dual machine learning pinpoints the Radius of Informative Structural Environments in metallic glasses. npj Comput Mater 12, 122 (2026). https://doi.org/10.1038/s41524-026-01997-z
Nyckelord: metalliskt glas, maskininlärning, atomär struktur, medelräckviddsordning, konfigurativ energi