Clear Sky Science · en
Dual machine learning pinpoints the Radius of Informative Structural Environments in metallic glasses
Why this hidden scale in glass matters
Metallic glasses are metals that have been cooled so quickly they never form a crystal. This lack of regular structure gives them unusual strength and toughness, but it also makes them hard to design on purpose: without a tidy crystal pattern, it has been unclear which parts of their atomic structure really control their behavior. This paper uses advanced machine learning to show that there is in fact a sweet-spot size in these disordered materials—a kind of “Goldilocks” neighborhood around each atom—where the structure contains the most useful information about how the whole material will act.
Seeing order in disordered metals
In ordinary crystalline metals, engineers can link strength and ductility to well-known features such as grain size or dislocations, which extend over many atoms in a regular lattice. Metallic glasses lack that long-range order and obvious defects, so researchers have tried to rely on more local building blocks called short-range orders, the way atoms pack around a central atom. Earlier work showed that these closest neighbors are not enough: the same local motif can behave very differently depending on its surroundings. That pointed to a key open question: over what distance around each atom do we need to look to capture the structural patterns that actually control bulk properties like strength and stability?
A top-down look at atomic neighborhoods
The authors first take a reductionist, or top-down, approach. They represent each atom’s surroundings as a mathematical fingerprint and group similar fingerprints into a set of unique local environments. For each metallic glass sample, they count how often each environment appears and train a machine learning model (XGBoost) to predict the sample’s total configurational energy, a quantity closely tied to how strong or ductile the material is. Crucially, they repeat this whole process while changing how far out from each atom they look—from just the first neighbors to distances spanning several neighbor shells. The model’s prediction error does not simply improve as they include more atoms. Instead, it reaches its best performance when the radius is about 5 ångströms, roughly out to the second-neighbor shell, and then worsens again. At that radius, the number and diversity of distinct local environments peak, and the spread in a structural entropy measure is widest, indicating that this scale packs in the richest structural information.
A bottom-up view from 3D images
To test whether this special radius might be an artifact of their first method, the team builds a second, very different model inspired by modern image recognition. They convert each atomic structure into a pair of three-dimensional pixel grids—one channel for each chemical species—and feed these into a vision transformer, a neural network that learns patterns in patches of the grid. By tuning how far these patches are allowed to “talk” to each other, they control an effective communication length that sets the largest structural scale the model can use. As they increase this length, the model’s accuracy improves but then levels off: beyond a communication length corresponding to a spherical radius of about 5 ångströms, additional range adds almost no benefit. Independent analysis of which regions in the grid most influence the prediction shows that the network’s attention also converges once this same scale is reached.
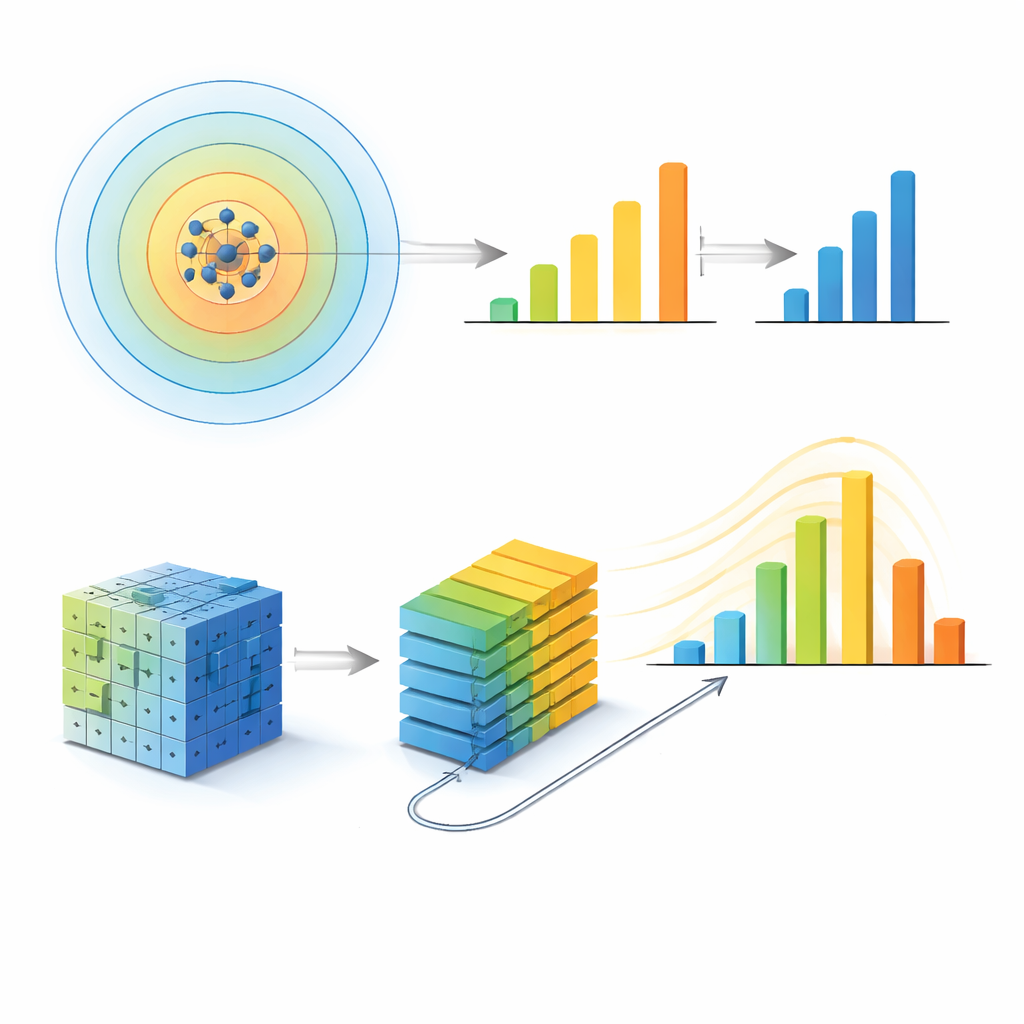
Robust across sizes and different glasses
The agreement between these two contrasting machine learning views—one built on hand-crafted atomic fingerprints, the other on raw voxel grids—suggests that the identified length scale is a real physical feature, not a modeling quirk. The authors further stress-test this idea by changing technical settings, system sizes, and even the types of glassy materials. Larger simulation boxes with more atoms preserve the same optimal radius. When they repeat the analysis for more complex metallic glasses containing aluminum, and for a chemically distinct palladium–silicon glass, each system again shows a specific informative radius, which is slightly smaller in the more tightly packed Pd–Si case. Similar behavior appears in amorphous carbon and silicon, where covalent bonds lead to different but still well-defined characteristic scales. Across all these cases, the radius where model performance peaks or saturates lines up with independent structural and experimental clues about medium-range organization in these materials.
What the study reveals in simple terms
For a non-specialist, the central message is that even in a seemingly random metallic glass, there is a natural “zone of influence” around each atom—about two atoms out—where the arrangement of neighbors carries almost all the information needed to predict how the entire piece of glass will behave energetically. Looking only at the very nearest neighbors misses important context, while looking too far out just adds redundant detail. By pinpointing this radius of informative structural environments, the work provides a practical target scale for future models and experiments. It offers a roadmap for designing better metallic glasses and other amorphous materials by focusing attention on the structural motifs that live at this intermediate scale, where hidden order in disorder most strongly shapes macroscopic properties.
Citation: Wang, M., Wang, Y., Islam, M. et al. Dual machine learning pinpoints the Radius of Informative Structural Environments in metallic glasses. npj Comput Mater 12, 122 (2026). https://doi.org/10.1038/s41524-026-01997-z
Keywords: metallic glass, machine learning, atomic structure, medium-range order, configurational energy