Clear Sky Science · it
Il machine learning duale individua il raggio degli ambienti strutturali informativi nei vetri metallici
Perché questa scala nascosta nel vetro è importante
I vetri metallici sono metalli raffreddati così rapidamente da non riuscire a formare un cristallo. Questa assenza di struttura regolare conferisce loro proprietà di resistenza e tenacità insolite, ma rende anche difficile progettarli intenzionalmente: senza un reticolo cristallino ordinato non è chiaro quali parti della loro struttura atomica controllino davvero il comportamento. Questo articolo usa tecniche avanzate di machine learning per dimostrare che esiste, in realtà, una dimensione ottimale in questi materiali disordinati — una sorta di «vicinato Goldilocks» attorno a ciascun atomo — in cui la struttura contiene le informazioni più utili per prevedere come si comporterà l’intero materiale.
Vedere ordine nei metalli disordinati
Nei metalli cristallini ordinari, gli ingegneri possono collegare resistenza e duttilità a caratteristiche note come la dimensione dei grani o le dislocazioni, che si estendono su molti atomi in un reticolo regolare. I vetri metallici non hanno quell’ordine a lungo raggio né difetti evidenti, perciò i ricercatori hanno cercato di fare riferimento a elementi locali più ridotti chiamati ordini a corto raggio, cioè il modo in cui gli atomi si dispongono attorno a un atomo centrale. Lavori precedenti hanno mostrato che questi vicini più prossimi non sono sufficienti: lo stesso motivo locale può comportarsi in modo molto diverso a seconda del contesto circostante. Questo ha portato a una domanda chiave: a quale distanza attorno a ciascun atomo bisogna guardare per cogliere i modelli strutturali che controllano davvero proprietà macroscopiche come resistenza e stabilità?
Uno sguardo dall’alto ai vicinati atomici
Gli autori adottano innanzitutto un approccio riduzionista, o dall’alto verso il basso. Rappresentano l’intorno di ciascun atomo come un'impronta matematica e raggruppano impronte simili in un insieme di ambienti locali distinti. Per ogni campione di vetro metallico, contano quanto spesso appare ciascun ambiente e addestrano un modello di machine learning (XGBoost) per prevedere l’energia configurazionale totale del campione, una grandezza strettamente legata a quanto il materiale sia resistente o duttile. Crucialmente, ripetono l’intero processo variando quanto lontano dal singolo atomo osservano — dai soli primi vicini fino a distanze che coprono diversi involucri di vicinato. L’errore di previsione del modello non migliora semplicemente includendo più atomi: raggiunge invece la migliore performance quando il raggio è di circa 5 ångström, approssimativamente fino al secondo shell di vicini, e poi peggiora nuovamente. A quel raggio, il numero e la diversità degli ambienti locali distinti raggiungono un massimo e la dispersione di una misura di entropia strutturale è più ampia, indicando che questa scala concentra le informazioni strutturali più ricche.
Una visione dal basso basata su immagini 3D
Per verificare se questo raggio speciale potesse essere un artefatto del primo metodo, il gruppo costruisce un secondo modello molto diverso ispirato al riconoscimento di immagini moderno. Convertono ciascuna struttura atomica in una coppia di griglie tridimensionali di pixel — un canale per ogni specie chimica — e le immettono in un vision transformer, una rete neurale che apprende pattern in patch della griglia. Regolando quanto queste patch sono autorizzate a «comunicare» tra loro, controllano una lunghezza di comunicazione effettiva che fissa la massima scala strutturale utilizzabile dal modello. Aumentando questa lunghezza, l’accuratezza del modello migliora ma poi si stabilizza: oltre una lunghezza di comunicazione corrispondente a un raggio sferico di circa 5 ångström, l’estensione ulteriore apporta quasi nessun beneficio. Un’analisi indipendente delle regioni della griglia che influenzano maggiormente la previsione mostra che anche l’attenzione della rete converge quando si raggiunge la stessa scala.
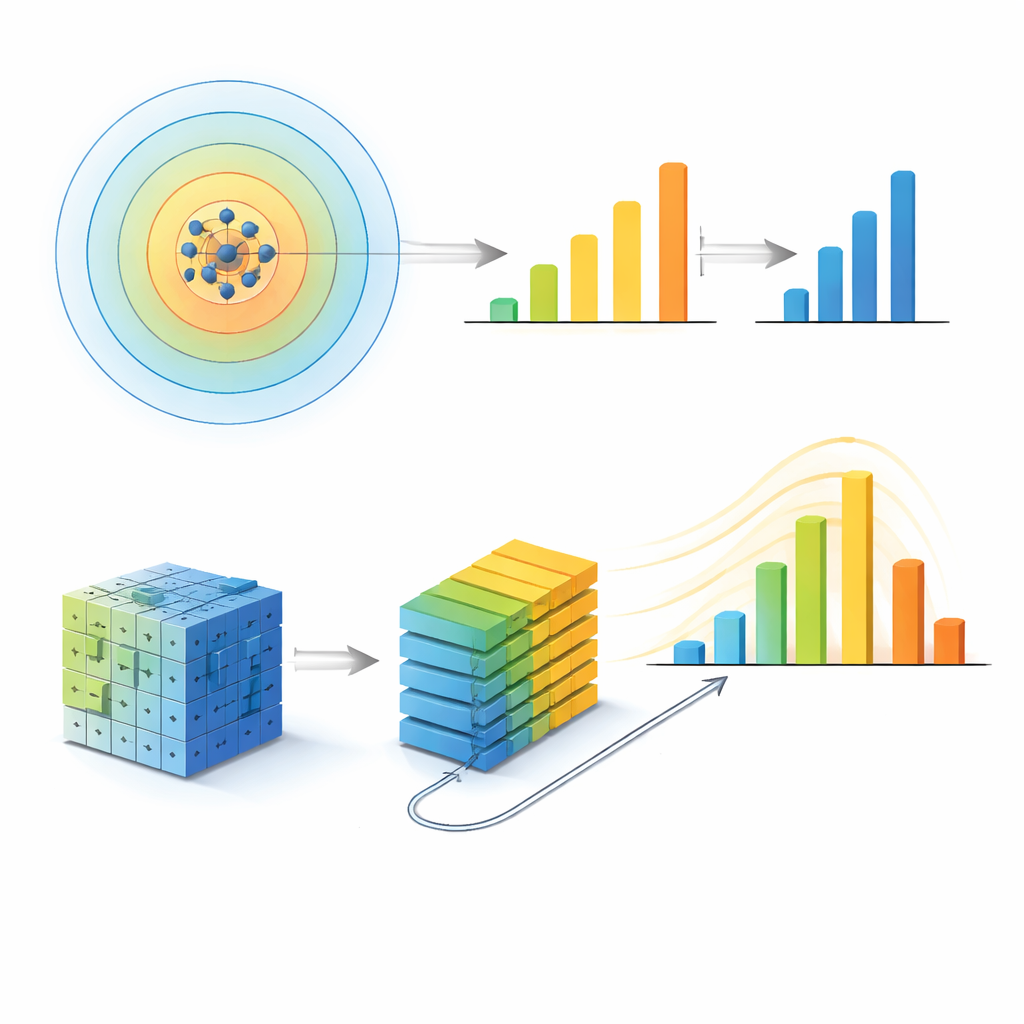
Robusto attraverso dimensioni e diversi vetri
L’accordo tra queste due visioni di machine learning contrastanti — una basata su impronte atomiche costruite a mano, l’altra su griglie di voxel grezze — suggerisce che la scala individuata sia una caratteristica fisica reale, non un artefatto del modello. Gli autori mettono ulteriormente alla prova questa idea variando parametri tecnici, le dimensioni dei sistemi e persino i tipi di materiali vetrosi. Cassette di simulazione più grandi con più atomi preservano lo stesso raggio ottimale. Ripetendo l’analisi per vetri metallici più complessi contenenti alluminio e per un vetro chimicamente distinto a base di palladio–silicio, ciascun sistema mostra di nuovo un raggio informativo specifico, leggermente più piccolo nel caso più compatto Pd–Si. Un comportamento simile si osserva in carbonio e silicio amorfi, dove i legami covalenti portano a scale caratteristiche diverse ma comunque ben definite. In tutti questi casi, il raggio in cui le prestazioni del modello raggiungono il picco o si saturano corrisponde a indizi strutturali ed esperimentali indipendenti sull’organizzazione a media distanza in questi materiali.
Cosa rivela lo studio in termini semplici
Per un pubblico non specialistico, il messaggio centrale è che anche in un vetro metallico apparentemente casuale esiste una naturale «zona di influenza» attorno a ciascun atomo — circa due atomi di distanza — in cui la disposizione dei vicini contiene quasi tutte le informazioni necessarie per prevedere come si comporterà energeticamente l’intero pezzo di vetro. Guardare soltanto i vicini immediati più prossimi manca di contesto importante, mentre guardare troppo lontano aggiunge solo dettagli ridondanti. Individuando questo raggio degli ambienti strutturali informativi, il lavoro fornisce una scala pratica per futuri modelli e esperimenti. Offre una road map per progettare vetri metallici e altri materiali amorfi migliorando l’attenzione sui motivi strutturali che abitano questa scala intermedia, dove l’ordine nascosto nel disordine influenza maggiormente le proprietà macroscopiche.
Citazione: Wang, M., Wang, Y., Islam, M. et al. Dual machine learning pinpoints the Radius of Informative Structural Environments in metallic glasses. npj Comput Mater 12, 122 (2026). https://doi.org/10.1038/s41524-026-01997-z
Parole chiave: vetro metallico, machine learning, struttura atomica, ordine a media distanza, energia configurazionale