Clear Sky Science · fr
Un apprentissage automatique double identifie le rayon des environnements structurels informatifs dans les verres métalliques
Pourquoi cette échelle cachée dans le verre compte
Les verres métalliques sont des métaux refroidis si rapidement qu’ils ne forment jamais de cristal. Cette absence de structure régulière leur confère une résistance et une ténacité inhabituelles, mais rend aussi leur conception difficile : sans un réseau cristallin ordonné, il est resté ambigues quelles parties de leur structure atomique contrôlent vraiment leur comportement. Cet article utilise des techniques avancées d’apprentissage automatique pour montrer qu’il existe en fait une taille optimale dans ces matériaux désordonnés — une sorte de voisinage « ni trop grand ni trop petit » autour de chaque atome — où la structure contient l’information la plus utile pour prédire le comportement de l’ensemble du matériau.
Voir de l’ordre dans des métaux désordonnés
Dans les métaux cristallins ordinaires, les ingénieurs peuvent lier résistance et ductilité à des caractéristiques bien connues comme la taille des grains ou les dislocations, qui s’étendent sur de nombreux atomes dans un réseau régulier. Les verres métalliques n’ont pas cet ordre à longue portée ni de défauts évidents, si bien que les chercheurs se sont appuyés sur des blocs plus locaux appelés ordres à courte portée, c’est‑à‑dire la façon dont les atomes s’organisent autour d’un atome central. Des travaux antérieurs ont montré que ces plus proches voisins ne suffisent pas : un même motif local peut se comporter très différemment selon son environnement. Cela soulève une question clé restée ouverte : sur quelle distance autour de chaque atome doit‑on regarder pour capturer les motifs structurels qui contrôlent effectivement des propriétés globales comme la résistance et la stabilité ?
Un regard descendante sur les voisinages atomiques
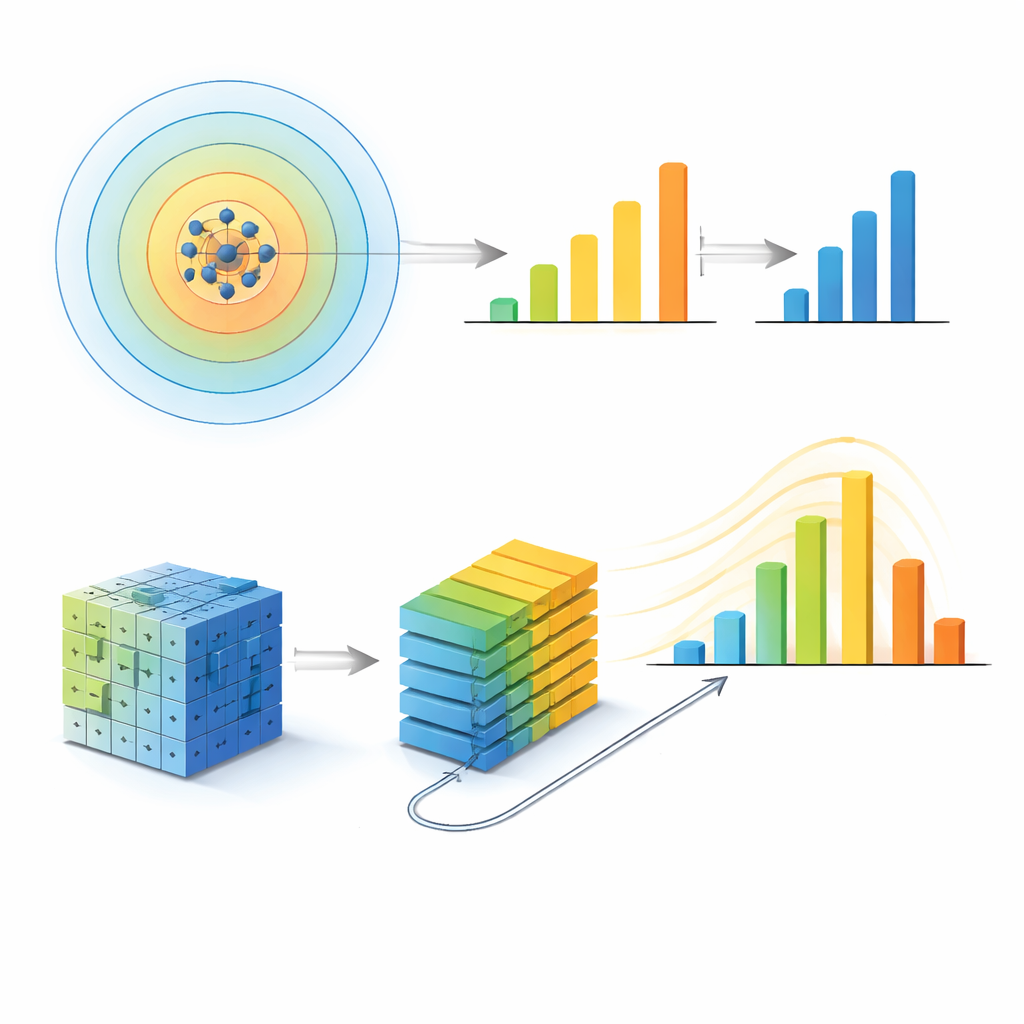
Les auteurs adoptent d’abord une approche réductionniste, ou descendante. Ils représentent l’entourage de chaque atome par une empreinte mathématique et regroupent les empreintes similaires en un ensemble d’environnements locaux uniques. Pour chaque échantillon de verre métallique, ils comptent la fréquence d’apparition de chaque environnement et entraînent un modèle d’apprentissage automatique (XGBoost) pour prédire l’énergie configurationnelle totale de l’échantillon, grandeur étroitement liée à la résistance ou à la ductilité du matériau. Crucialement, ils répètent l’ensemble du processus en changeant la distance considérée autour de chaque atome — depuis les seuls premiers voisins jusqu’à des distances couvrant plusieurs couches de voisins. L’erreur de prédiction du modèle ne s’améliore pas simplement en incluant davantage d’atomes. Au contraire, elle atteint ses meilleures performances pour un rayon d’environ 5 ångströms, soit à peu près la deuxième couche de voisins, puis se dégrade de nouveau. À ce rayon, le nombre et la diversité des environnements locaux distincts atteignent un maximum, et la dispersion d’une mesure d’entropie structurelle est la plus large, indiquant que cette échelle concentre l’information structurelle la plus riche.
Une vue ascendante à partir d’images 3D
Pour vérifier si ce rayon particulier n’était pas un artéfact de leur première méthode, l’équipe construit un second modèle très différent, inspiré de la reconnaissance d’images moderne. Ils convertissent chaque structure atomique en une paire de grilles de pixels tridimensionnelles — une couche par espèce chimique — et les injectent dans un vision transformer, un réseau de neurones qui apprend des motifs dans des patches de la grille. En réglant la distance à laquelle ces patches peuvent « communiquer », ils contrôlent une longueur de communication effective qui fixe l’échelle structurelle maximale dont le modèle peut tirer parti. En augmentant cette longueur, la précision du modèle s’améliore puis se stabilise : au‑delà d’une longueur de communication correspondant à un rayon sphérique d’environ 5 ångströms, une portée supplémentaire apporte presque aucun bénéfice. Une analyse indépendante des régions de la grille qui influencent le plus la prédiction montre que l’attention du réseau converge également une fois cette même échelle atteinte.
Robuste selon la taille et les différents verres
L’accord entre ces deux vues d’apprentissage automatique contrastées — l’une fondée sur des empreintes atomiques fabriquées à la main, l’autre sur des voxel bruts — suggère que l’échelle mise en évidence est une caractéristique physique réelle, et non un artefact de modélisation. Les auteurs mettent cette idée à l’épreuve en changeant des paramètres techniques, la taille des systèmes et même les types de matériaux vitreux. Des boîtes de simulation plus grandes avec davantage d’atomes conservent le même rayon optimal. Lorsqu’ils répètent l’analyse pour des verres métalliques plus complexes contenant de l’aluminium, et pour un verre palladium–silicium chimiquement distinct, chaque système affiche de nouveau un rayon informatif spécifique, légèrement plus petit dans le cas plus densément empaqueté Pd–Si. Un comportement similaire apparaît dans le carbone et le silicium amorphes, où les liaisons covalentes conduisent à des échelles caractéristiques différentes mais tout aussi bien définies. Dans tous ces cas, le rayon où la performance du modèle atteint un pic ou se sature coïncide avec des indices structurels et expérimentaux indépendants sur l’organisation à moyenne portée dans ces matériaux.
Ce que l’étude révèle en termes simples
Pour un non‑spécialiste, le message central est que même dans un verre métallique apparemment aléatoire, il existe une « zone d’influence » naturelle autour de chaque atome — d’environ deux atomes de rayon — où l’agencement des voisins contient presque toute l’information nécessaire pour prédire le comportement énergétique de la pièce de verre dans son ensemble. Ne considérer que les plus proches voisins immédiats fait manquer un contexte important, tandis que regarder trop loin n’ajoute que des détails redondants. En identifiant ce rayon des environnements structurels informatifs, le travail fournit une échelle pratique pour les futurs modèles et expériences. Il propose une feuille de route pour concevoir de meilleurs verres métalliques et autres matériaux amorphes en concentrant l’attention sur les motifs structurels qui résident à cette échelle intermédiaire, où l’ordre caché dans le désordre influence le plus fortement les propriétés macroscopiques.
Citation: Wang, M., Wang, Y., Islam, M. et al. Dual machine learning pinpoints the Radius of Informative Structural Environments in metallic glasses. npj Comput Mater 12, 122 (2026). https://doi.org/10.1038/s41524-026-01997-z
Mots-clés: verre métallique, apprentissage automatique, structure atomique, ordre à moyenne portée, énergie configurationnelle