Clear Sky Science · sv
Hämning av PTBP1 omprogrammerar myogenes för att rädda försämrad muskelregeneration hos mdx‑möss genom att korrigera E2A‑splitsning
Varför det spelar roll att laga trötta muskler
Duchenne muskeldystrofi är en förödande barndomssjukdom där muskler gradvis försvagas och förtvinar. Medan många experimentella behandlingar försöker ersätta det saknade dystrofinproteinet ställer den här studien en annan fråga: kan vi få kroppens egna muskelstamceller att bygga upp starkare vävnad, även när genfelet kvarstår? Författarna identifierar en molekylär växel som talar om för muskelceller när de ska dela sig och när de ska mogna, och de visar att att återställa balansen i denna växel kan återuppliva muskelreparation i en musmodell för Duchenne.
Problemet med fastlåst muskelläkning
Friska muskler reparerar sig ständigt. Efter skada vaknar vilande stamceller, delar sig och smälter sedan samman till nya muskeltrådar. Vid Duchenne muskeldystrofi bryts denna cykel: musklerna är inflammerade, fibrerna degenererar kontinuerligt och försöken till regeneration hinner inte med. Vid undersökning av muskelprover från pojkar med Duchenne och från mdx‑möss — en standard djurmodell — såg forskarna många prolifererande muskelprekursorceller men påfallande få nya, funktionella fibrer. Blodmarkörer för muskelskada var höga och skadade områden ersattes av ärrvävnad och fett, vilket pekar på ett system fast i tillväxtfasen som misslyckas med att genomföra verklig reparation. 
En molekylär spak mellan tillväxt och mognad
Teamet fokuserade på en huvudregulatorprotein kallad E2A, som förekommer i två former, E12 och E47, framställda från samma gen genom alternativ splitsning — en process som klipper och sätter ihop RNA på olika sätt. I laboratorieodlade myoblaster dominerade E47 när cellerna snabbt delade sig, medan E12 ökade när cellerna började smälta samman och mogna till muskeltrådar. Genom att selektivt öka eller minska varje form visade författarna att E47 driver cellproliferation, medan E12 är nödvändig för att slå på det genetiska programmet för muskeldifferentiering. I muskler från Duchenne‑patienter och mdx‑möss var denna balans dock störd: totala E2A‑nivåer var höga, men E47‑varianten dominerade och E12‑varianten var relativt sällsynt, vilket speglade det observerade överskottet av tillväxt utan korrekt mognad.
Splitsningsväktaren PTBP1
För att ta reda på vad som tippar balansen mellan E47 och E12 grävde forskarna i offentliga genuttrycksdatabaser och patientbiopsier efter splitsningsregulatorer som ändras under normal muskelutveckling och vid Duchenne. Ett protein, PTBP1, utmärkte sig. I friska myoblaster är PTBP1‑nivåerna höga under proliferation för att sedan falla kraftigt när cellerna börjar differentieras, vilket sammanfaller med skiftet från E47 till E12. I Duchenne‑muskler förblev PTBP1 onormalt förhöjt — mer än åtta gånger högre än hos opåverkade kontroller — och var koncentrerat i prolifererande prekursorer och även i små, omogna fibrer. I cellkultur och i skadad muskel ökade experimentellt förhöjd PTBP1 halten av celler i ett tillväxttillstånd, gynnade E47‑formen av E2A, blockerade bildandet av robusta nya fibrer och drev i slutändan vissa prekursorceller till celldöd. Att reducera PTBP1 gav motsatt effekt: det ökade E12, förbättrade differentieringsmarkörer och påskyndade framträdandet av nya fibrer vid skadeställen.
Rädda sjuka muskler genom att återställa växeln
Det avgörande testet var om finjustering av PTBP1 kunde förbättra muskeln hos mdx‑möss som modellerar Duchenne. Med en riktad genterapivektor som agerar specifikt i aktiverade muskelprekursorer slog teamet ned PTBP1 i dessa möss. Behandlade djur visade bättre muskelfunktion i häng‑ och griptester, lägre blodmarkörer för skada och fler och större regenererande fibrer i flera muskler, inklusive diafragman. Molekylära analyser bekräftade att PTBP1‑nedsättning skiftade E2A‑splitsningen mot E12‑formen, minskade proliferationsmarkören MyoD och ökade differentieringssignaler och tidiga muskel‑fibrerproteiner, vilket indikerar att prekursorcellerna slutligen fullföljde sin utvecklingsresa.
En läkemedelsstrategi för att ta bort bromsen
Eftersom det är utmanande att direkt rikta in sig på PTBP1 med designade läkemedel undersökte forskarna uppströms hur proteinet bryts ner. De identifierade ett deubiquitinase‑enzym, USP9X, som normalt skyddar PTBP1 från nedbrytning. I Duchenne‑modeller var USP9X onormalt högt. Behandling av mdx‑möss med degrasyn, en småmolekylär hämmare av deubiquitinaser inklusive USP9X, minskade PTBP1‑proteinnivåerna, ökade differentieringsmarkören Myogenin och förbättrade muskelstruktur och funktion. Muskelfibrer var mindre nekrotiska, blodmarkörer för skada sjönk och gång och hängprestanda förbättrades, vilket tyder på att farmakologiskt tryck mot PTBP1‑stabilitet kan återställa det regenerativa programmet. 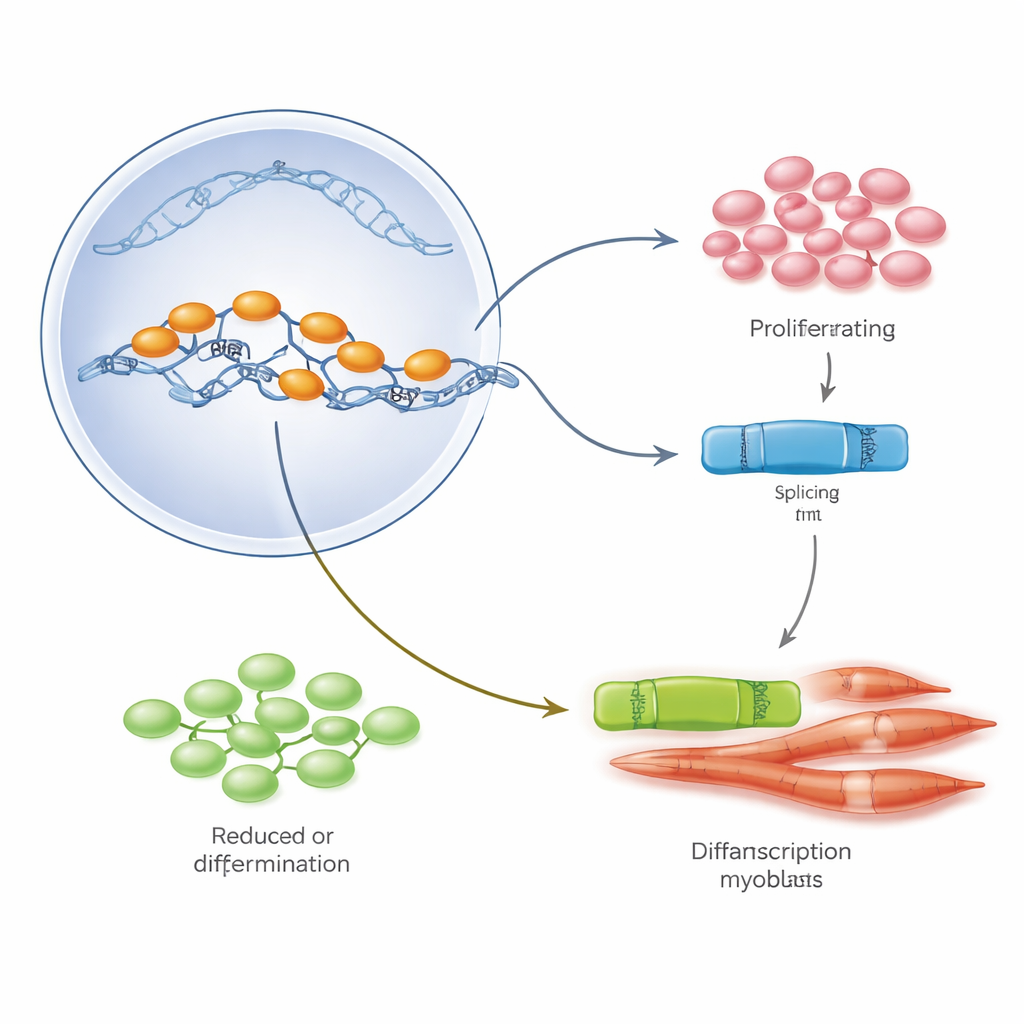
Vad detta kan innebära för patienter
Denna studie visar att, utöver den saknade dystrofin, är Duchenne‑muskler fångade i en felaktig utvecklingsloop: stamceller fortsätter cykla men blir inte mogna fibrer. PTBP1–E2A‑splitsningssystemet fungerar som en spak mellan tillväxt och differentiering. Vid Duchenne är den spaken fastlåst mot ändlös proliferation genom höga PTBP1‑nivåer och ett överskott av E47. Genom att sänka PTBP1 — genetiskt eller med ett läkemedel som degrasyn — kan spaken återställas och gynna E12‑formen som driver korrekt muskelbildning. Mycket återstår innan detta kan testas på barn, men studien utpekar en lovande ny väg för att hjälpa muskler att bygga upp sig själva, potentiellt i komplement till genersättningsbehandlingar och befintliga steroidbehandlingar.
Citering: Fan, S., Liu, X., Pan, Q. et al. PTBP1 inhibition reprograms myogenesis to rescue impaired muscle regeneration in mdx mice through correcting E2A splicing. Nat Commun 17, 3838 (2026). https://doi.org/10.1038/s41467-026-70669-9
Nyckelord: Duchenne muskeldystrofi, muskelregeneration, alternativ splitsning, PTBP1, mdx‑mus