Clear Sky Science · pl
Hamowanie PTBP1 przekierowuje miogenezę, przywracając zaburzoną regenerację mięśni u myszy mdx przez korektę splicingu E2A
Dlaczego naprawa zmęczonych mięśni ma znaczenie
Dystrofia mięśniowa Duchenne’a to wyniszczająca choroba dziecięca, w której mięśnie stopniowo słabną i zanikają. Wiele eksperymentalnych terapii próbuje zastąpić brakujące białko dystrofinę; w tej pracy zadano jednak inne pytanie: czy można nakłonić własne komórki macierzyste mięśni do odbudowy silniejszej tkanki, nawet jeśli defekt genetyczny pozostaje? Autorzy opisują molekularny przełącznik, który mówi komórkom mięśniowym, kiedy się mnożyć, a kiedy dojrzewać, i pokazują, że przywrócenie równowagi tego przełącznika może ożywić naprawę mięśni w modelu myszy choroby Duchenne’a.
Problem zatrzymanej regeneracji mięśni
Zdrowe mięśnie nieustannie się naprawiają. Po urazie uśpione komórki macierzyste budzą się, mnożą, a następnie łączą w nowe włókna mięśniowe. W dystrofii Duchenne’a ten cykl się załamuje: mięśnie są zapalne, włókna ciągle ulegają degeneracji, a próby regeneracji nie nadążają. Analiza próbek mięśni od chłopców z Duchenne’em i od myszy mdx — standardowego modelu zwierzęcego — ujawniła wiele proliferujących komórek prekursorowych mięśnia, ale zaskakująco niewiele nowych, funkcjonalnych włókien. Markery krwi świadczące o uszkodzeniu mięśni były podwyższone, a uszkodzone obszary zastępowane blizną i tłuszczem, co wskazuje na system utkwiony w fazie wzrostu i niezdolny do dokończenia prawdziwej naprawy. 
Molekularna dźwignia między wzrostem a dojrzewaniem
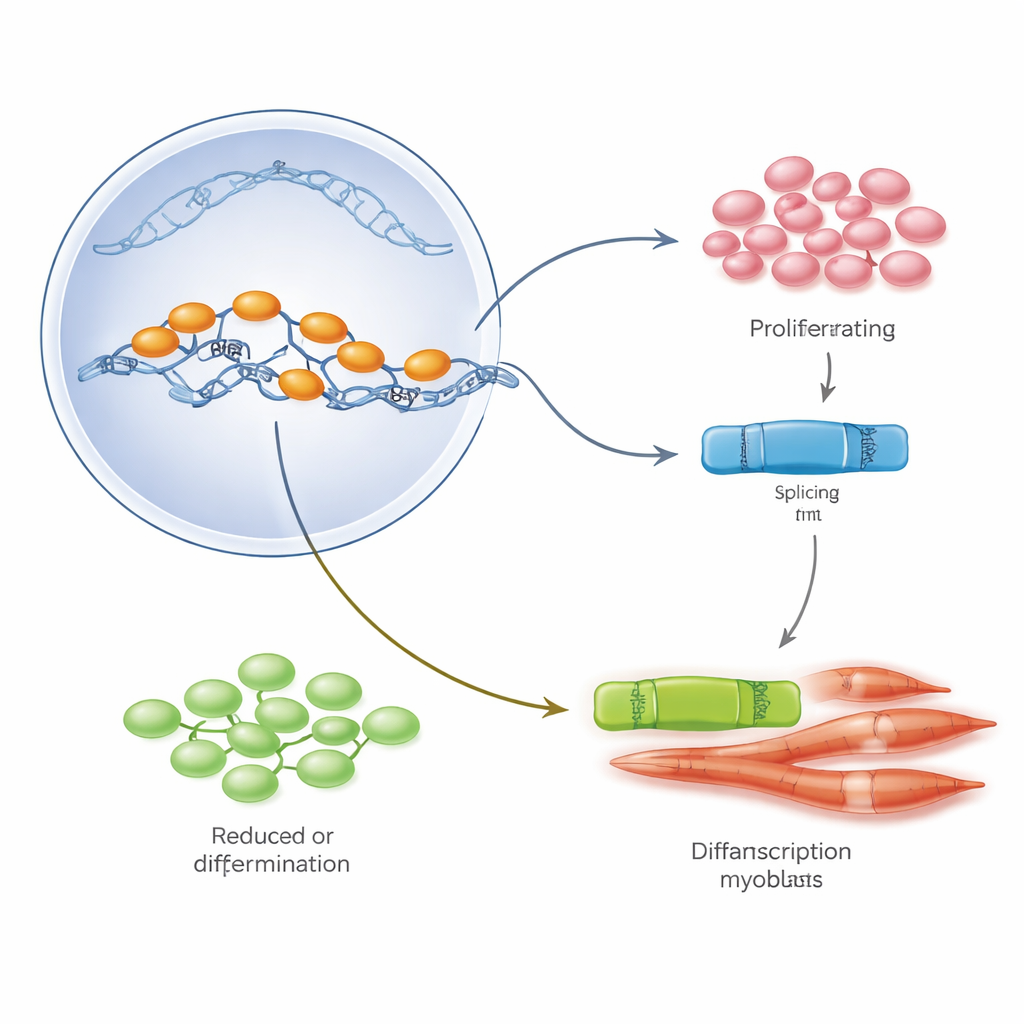
Zespół skupił się na głównym regulatorze białkowym zwanym E2A, który występuje w dwóch formach, E12 i E47, powstających z tego samego genu poprzez alternatywny splicing — proces wycinania i łączenia RNA na różne sposoby. W hodowlach mioblastów dominował E47 gdy komórki intensywnie się dzieliły, natomiast E12 wzrastał w miarę, jak komórki zaczynały się łączyć i dojrzewać w włókna mięśniowe. Poprzez selektywne zwiększanie lub zmniejszanie poziomów każdej formy, autorzy wykazali, że E47 napędza proliferację komórek, podczas gdy E12 jest niezbędny do uruchomienia programu genetycznego różnicowania mięśni. W mięśniach pacjentów z Duchenne’em i u myszy mdx równowaga ta była jednak zaburzona: całkowite poziomy E2A były wysokie, ale przeważała wersja E47, a E12 było stosunkowo mało, co odzwierciedla obserwowany nadmiar wzrostu bez prawidłowego dojrzewania.
Stróż splicingu — PTBP1
Aby sprawdzić, co przechyla szalę między E47 a E12, badacze przeszukali publiczne zestawy danych ekspresji genów i biopsje pacjentów w poszukiwaniu regulatorów splicingu, które zmieniają się podczas normalnego rozwoju mięśni i w chorobie Duchenne’a. Jedno białko, PTBP1, wyróżniało się. W zdrowych mioblastach poziomy PTBP1 są wysokie podczas proliferacji komórek, a następnie gwałtownie spadają na początku różnicowania, co pokrywa się ze zmianą z E47 na E12. W mięśniach z Duchenne’em PTBP1 pozostawał nieprawidłowo podwyższony — ponad ośmiokrotnie wyższy niż u kontrolnych osobników — i był skoncentrowany w proliferujących prekursorach, a nawet w małych, niedojrzałych włóknach. W hodowli komórkowej i w uszkodzonych mięśniach myszy eksperymentalne zwiększenie PTBP1 utrzymywało komórki w stanie wzrostu, promowało formę E47 E2A, blokowało tworzenie silnych nowych włókien i w końcu prowadziło do śmierci części komórek prekursorowych. Zmniejszenie PTBP1 dawało efekt odwrotny: podnosiło E12, zwiększało markery różnicowania i przyspieszało pojawienie się nowych włókien w miejscach urazu.
Ratowanie chorych mięśni przez przywrócenie równowagi przełącznika
Kluczowy test polegał na sprawdzeniu, czy regulacja PTBP1 może poprawić stan mięśni u myszy mdx, modelujących Duchenne’a. Przy użyciu ukierunkowanego wektora terapii genowej działającego specyficznie w aktywowanych prekursorach mięśniowych, zespół wyciszył PTBP1 u tych myszy. Zwierzęta po leczeniu wykazały lepszą wydolność mięśni w testach wiszenia i chwytu, niższe markery uszkodzenia we krwi oraz więcej i większe regenerujące się włókna w kilku mięśniach, w tym w przeponie. Analizy molekularne potwierdziły, że wyciszenie PTBP1 przesunęło splicing E2A w stronę formy E12, zmniejszyło marker proliferacji MyoD oraz zwiększyło sygnały różnicowania i wczesne białka włókien mięśniowych, co wskazuje, że komórki prekursorowe wreszcie kończyły swoją drogę rozwojową.
Strategia lekowa do zdjęcia hamulca
Ponieważ bezpośrednie celowanie w PTBP1 przy pomocy projektowanych leków jest trudne, badacze spojrzeli w górę szlaku, na mechanizmy degradacji tego białka. Zidentyfikowali enzym deubikwitynujący, USP9X, który normalnie chroni PTBP1 przed rozkładem. W modelach Duchenne’a USP9X był nieprawidłowo wysoki. Leczenie myszy mdx degrasyną, małocząsteczkowym inhibitorem deubikwitynaz, w tym USP9X, obniżyło poziomy białka PTBP1, zwiększyło marker różnicowania Myogenin i poprawiło strukturę oraz funkcję mięśni. Włókna mięśniowe były mniej nekrotyczne, markery uszkodzenia spadły, a chód i wydolność w teście wiszenia uległy poprawie, sugerując, że farmakologiczne skłonienie PTBP1 ku degradacji może przywrócić program regeneracyjny.
Co to może oznaczać dla pacjentów
Ta praca ujawnia, że poza brakującą dystrofiną mięśnie w Duchenne’ie utknęły w wadliwym pętlowym stanie rozwojowym: komórki macierzyste ciągle się dzielą, ale nie przekształcają w dojrzałe włókna. System splicingu PTBP1–E2A działa jak dźwignia między wzrostem a różnicowaniem. W Duchenne’ie dźwignia jest zablokowana w kierunku niekończącej się proliferacji z powodu wysokiego PTBP1 i nadmiaru E47. Obniżając PTBP1 — genetycznie lub przy użyciu leku takiego jak degrasyna — można przesunąć dźwignię z powrotem, faworyzując formę E12, która napędza prawidłowe tworzenie mięśni. Chociaż wiele pozostaje do zrobienia, zanim podejście to zostanie przetestowane u dzieci, badanie wyznacza obiecującą nową drogę do wspierania zdolności mięśni do samoodbudowy, potencjalnie uzupełniając terapie zastępcze genów i istniejące leczenie steroidami.
Cytowanie: Fan, S., Liu, X., Pan, Q. et al. PTBP1 inhibition reprograms myogenesis to rescue impaired muscle regeneration in mdx mice through correcting E2A splicing. Nat Commun 17, 3838 (2026). https://doi.org/10.1038/s41467-026-70669-9
Słowa kluczowe: Dystrofia mięśniowa Duchenne’a, regeneracja mięśni, splicing alternatywny, PTBP1, mysz mdx