Clear Sky Science · fr
L’inhibition de PTBP1 reprogramme la myogenèse pour restaurer la régénération musculaire altérée chez les souris mdx en corrigeant l’épissage de E2A
Pourquoi réparer des muscles fatigués compte
La dystrophie musculaire de Duchenne est une maladie infantile dévastatrice dans laquelle les muscles s’affaiblissent et s’atrophient progressivement. Alors que de nombreux traitements expérimentaux visent à remplacer la protéine dystrophine manquante, cet article pose une question différente : peut-on inciter les propres cellules souches musculaires du corps à reconstruire un tissu plus robuste, même si le défaut génétique persiste ? Les auteurs mettent au jour un interrupteur moléculaire qui indique aux cellules musculaires quand se multiplier et quand mûrir, et montrent que rééquilibrer cet interrupteur peut raviver la réparation musculaire dans un modèle murin de Duchenne.
Le problème d’une réparation musculaire bloquée
Les muscles sains se réparent en permanence. Après une lésion, des cellules souches dormantes se réveillent, se multiplient, puis fusionnent pour former de nouvelles fibres musculaires. Dans la dystrophie de Duchenne, ce cycle se dérègle : les muscles sont enflammés, les fibres dégénèrent en continu, et les tentatives de régénération n’arrivent pas à suivre. En examinant des échantillons musculaires provenant de garçons atteints de Duchenne et de souris mdx—un modèle animal standard—les chercheurs ont observé de nombreuses cellules précurseures en prolifération mais étonnamment peu de nouvelles fibres fonctionnelles. Les marqueurs sanguins de dommage musculaire étaient élevés, et les zones lésées du muscle étaient remplacées par de la cicatrice et de la graisse, autant d’indices d’un système coincé en phase de croissance et incapable d’achever une véritable réparation. 
Un levier moléculaire entre croissance et maturation
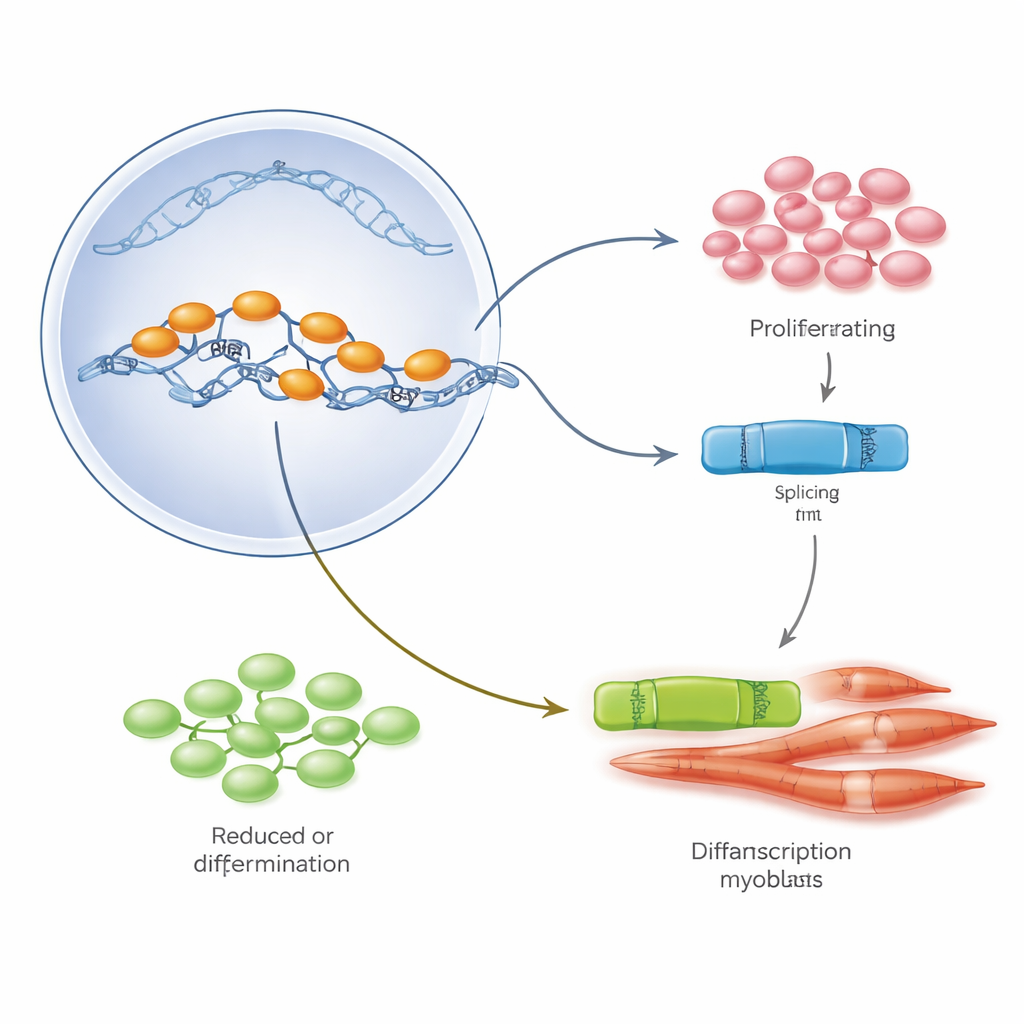
L’équipe s’est concentrée sur une protéine régulatrice maîtresse appelée E2A, qui existe sous deux formes, E12 et E47, générées à partir du même gène par épissage alternatif, un processus qui découpe et recolle l’ARN de manières différentes. Dans des myoblastes en culture, E47 dominait lorsque les cellules se divisaient rapidement, tandis que E12 augmentait quand les cellules commençaient à fusionner et à mûrir en fibres musculaires. En modulant sélectivement chacune des formes, les auteurs ont montré qu’E47 alimente la prolifération cellulaire, tandis qu’E12 est essentielle pour activer le programme génétique de différenciation musculaire. Dans les muscles de patients Duchenne et chez les souris mdx, cependant, cet équilibre était perturbé : les niveaux totaux d’E2A étaient élevés, mais la version E47 prédominait et la version E12 était relativement rare, reflétant l’excès de croissance observé sans maturation appropriée.
Le gardien de l’épissage PTBP1
Pour savoir ce qui fait pencher la balance entre E47 et E12, les chercheurs ont exploré des jeux de données publics d’expression génique et des biopsies de patients à la recherche de régulateurs d’épissage qui changent au cours du développement musculaire normal et dans la maladie de Duchenne. Une protéine, PTBP1, est sortie du lot. Dans les myoblastes sains, les niveaux de PTBP1 sont élevés pendant la prolifération, puis chutent fortement au début de la différenciation, coïncidant avec le passage d’E47 à E12. Dans le muscle Duchenne, PTBP1 restait anormalement élevé—plus de huit fois supérieur aux témoins non affectés—et se concentrait dans les précurseurs proliférants et même dans de petites fibres immatures. En culture cellulaire et dans le muscle lésé de souris, augmenter expérimentalement PTBP1 maintenait les cellules bloquées dans un état de croissance, favorisait la forme E47 d’E2A, empêchait la formation de nouvelles fibres robustes et conduisait finalement certains précurseurs à mourir. Réduire PTBP1 produisait l’effet inverse : cela augmentait E12, renforçait les marqueurs de différenciation et accélèrait l’apparition de nouvelles fibres sur les sites de lésion.
Sauver le muscle malade en rééquilibrant l’interrupteur
Le test crucial fut de savoir si moduler PTBP1 pouvait améliorer le muscle chez les souris mdx qui modélisent la Duchenne. Utilisant un vecteur de thérapie génique ciblé qui agit spécifiquement dans les précurseurs musculaires activés, l’équipe a réduit l’expression de PTBP1 chez ces souris. Les animaux traités ont montré de meilleures performances musculaires aux tests de suspension et de préhension, des marqueurs sanguins de dommage plus faibles, et davantage de fibres régénérantes plus grandes dans plusieurs muscles, y compris le diaphragme. Les analyses moléculaires ont confirmé que l’inhibition de PTBP1 décalait l’épissage d’E2A vers la forme E12, réduisait le marqueur de prolifération MyoD et augmentait les signaux de différenciation et les protéines précoces des fibres musculaires, indiquant que les cellules précurseures achevaient enfin leur parcours développemental.
Une stratégie médicamenteuse pour lever le frein
Parce qu’il est difficile de cibler directement PTBP1 avec des médicaments conçus sur mesure, les chercheurs ont regardé en amont la manière dont la protéine est dégradée. Ils ont identifié une déubiquitinase, USP9X, qui protège normalement PTBP1 de la dégradation. Dans les modèles de Duchenne, USP9X était anormalement élevé. Traiter des souris mdx avec degrasyn, une petite molécule inhibitrice de déubiquitinases incluant USP9X, a réduit les niveaux de protéine PTBP1, augmenté le marqueur de différenciation Myogénine et amélioré la structure et la fonction musculaires. Les fibres étaient moins nécrotiques, les marqueurs sanguins de dommage ont diminué, et la démarche ainsi que les performances aux tests de suspension se sont améliorées, suggérant que pousser pharmacologiquement PTBP1 vers la destruction peut restaurer le programme régénératif.
Ce que cela pourrait signifier pour les patients
Ce travail révèle que, au‑delà de la dystrophine manquante, les muscles Duchenne sont piégés dans une boucle développementale défaillante : les cellules souches continuent de se diviser mais ne deviennent pas des fibres matures. Le système d’épissage PTBP1–E2A agit comme un levier entre croissance et différenciation. Dans la Duchenne, ce levier est bloqué vers une prolifération sans fin par un PTBP1 élevé et un excès d’E47. En abaissant PTBP1—génétiquement ou avec un médicament comme degrasyn—on peut repousser le levier en faveur de la forme E12 qui pilote la formation musculaire correcte. Bien qu’il reste beaucoup à faire avant de tester cette approche chez l’enfant, l’étude propose une voie prometteuse pour aider les muscles à se reconstruire eux‑mêmes, en complément potentiel des thérapies de remplacement génique et des traitements stéroïdiens existants.
Citation: Fan, S., Liu, X., Pan, Q. et al. PTBP1 inhibition reprograms myogenesis to rescue impaired muscle regeneration in mdx mice through correcting E2A splicing. Nat Commun 17, 3838 (2026). https://doi.org/10.1038/s41467-026-70669-9
Mots-clés: Dystrophie musculaire de Duchenne, régénération musculaire, épissage alternatif, PTBP1, souris mdx