Clear Sky Science · en
PTBP1 inhibition reprograms myogenesis to rescue impaired muscle regeneration in mdx mice through correcting E2A splicing
Why fixing tired muscles matters
Duchenne muscular dystrophy is a devastating childhood disease in which muscles progressively weaken and waste away. While many experimental treatments try to replace the missing dystrophin protein, this paper asks a different question: can we coax the body’s own muscle stem cells to rebuild stronger tissue, even when the gene defect remains? The authors uncover a molecular switch that tells muscle cells when to multiply and when to mature, and they show that flipping this switch back into balance can revive muscle repair in a mouse model of Duchenne.
The problem of stalled muscle repair
Healthy muscles constantly repair themselves. After injury, dormant stem cells wake up, multiply, and then fuse into new muscle fibers. In Duchenne muscular dystrophy, this cycle breaks down: muscles are inflamed, fibers continuously degenerate, and attempts at regeneration cannot keep up. Examining muscle samples from boys with Duchenne and from mdx mice—a standard animal model—the researchers saw many proliferating muscle precursor cells but surprisingly few new, functional fibers. Blood markers of muscle damage were high, and damaged areas of muscle were being replaced by scar and fat, all pointing to a system stuck in the growth phase and failing to complete true repair. 
A molecular lever between growth and maturation
The team focused on a master regulator protein called E2A, which comes in two forms, E12 and E47, generated from the same gene by alternative splicing, a process that cuts and pastes RNA in different ways. In laboratory myoblasts, E47 dominated when cells were rapidly dividing, whereas E12 rose as cells began to fuse and mature into muscle fibers. By selectively dialing each form up or down, the authors showed that E47 fuels cell proliferation, while E12 is essential for turning on the genetic program of muscle differentiation. In Duchenne patient muscle and mdx mice, however, this balance was distorted: total E2A levels were high, but the E47 version predominated and the E12 version was relatively scarce, mirroring the observed excess of growth without proper maturation.
The splicing gatekeeper PTBP1
To find out what tips the scale between E47 and E12, the researchers mined public gene-expression datasets and patient biopsies for splicing regulators that change during normal muscle development and in Duchenne disease. One protein, PTBP1, stood out. In healthy myoblasts, PTBP1 levels are high while cells are proliferating, then fall sharply as they start to differentiate, coinciding with the shift from E47 to E12. In Duchenne muscle, PTBP1 remained abnormally elevated—more than eightfold higher than in unaffected controls—and was concentrated in proliferating precursors and even in small, immature fibers. In cell culture and in injured mouse muscle, experimentally increasing PTBP1 kept cells locked in a growth state, favored the E47 form of E2A, blocked formation of robust new fibers, and eventually drove some precursor cells to die. Reducing PTBP1 had the opposite effect: it boosted E12, enhanced differentiation markers, and sped the appearance of new fibers at injury sites.
Rescuing diseased muscle by rebalancing the switch
The crucial test was whether tuning PTBP1 could improve muscle in mdx mice that model Duchenne. Using a targeted gene therapy vector that acts specifically in activated muscle precursors, the team knocked down PTBP1 in these mice. Treated animals showed better muscle performance in hanging and grip tests, lower blood markers of damage, and more and larger regenerating fibers across several muscles, including the diaphragm. Molecular analyses confirmed that PTBP1 knockdown shifted E2A splicing toward the E12 form, reduced the proliferation marker MyoD, and raised differentiation signals and early muscle-fiber proteins, indicating that precursor cells were finally completing their developmental journey.
A drug strategy to clear the brake
Because directly targeting PTBP1 with designer drugs is challenging, the researchers looked upstream at how the protein is broken down. They identified a deubiquitinase enzyme, USP9X, that normally protects PTBP1 from degradation. In Duchenne models, USP9X was abnormally high. Treating mdx mice with degrasyn, a small-molecule inhibitor of deubiquitinases including USP9X, reduced PTBP1 protein levels, increased the differentiation marker Myogenin, and improved muscle structure and function. Muscle fibers were less necrotic, blood markers of damage fell, and gait and hanging performance improved, suggesting that pharmacologically nudging PTBP1 toward destruction can restore the regenerative program. 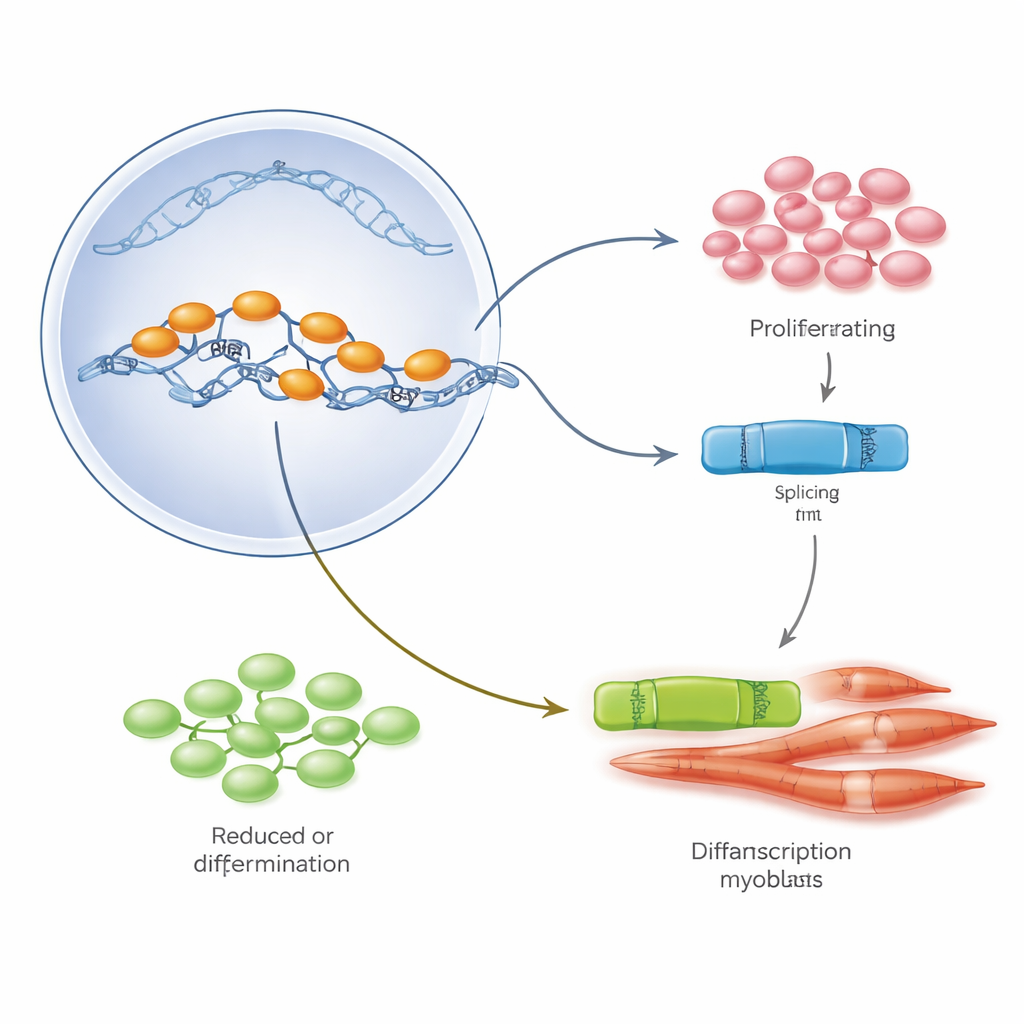
What this could mean for patients
This work reveals that, beyond the missing dystrophin, Duchenne muscles are trapped in a faulty developmental loop: stem cells keep cycling but fail to become mature fibers. The PTBP1–E2A splicing system acts as a lever between growth and differentiation. In Duchenne, that lever is jammed toward endless proliferation through high PTBP1 and excess E47. By lowering PTBP1—genetically or with a drug like degrasyn—the lever can be pushed back, favoring the E12 form that drives proper muscle formation. While much remains to be done before this approach can be tested in children, the study outlines a promising new way to help muscles rebuild themselves, potentially complementing gene-replacement therapies and existing steroid treatments.
Citation: Fan, S., Liu, X., Pan, Q. et al. PTBP1 inhibition reprograms myogenesis to rescue impaired muscle regeneration in mdx mice through correcting E2A splicing. Nat Commun 17, 3838 (2026). https://doi.org/10.1038/s41467-026-70669-9
Keywords: Duchenne muscular dystrophy, muscle regeneration, alternative splicing, PTBP1, mdx mouse