Clear Sky Science · sv
En meta-inlärnings- och uppgiftsanpassad metod för att förutsäga läkemedelsmålsaffinitet
Lära datorer att välja bättre läkemedel
Att hitta nya läkemedel innebär ofta att testa miljontals möjliga molekyler för att se vilka som binder till rätt proteiner i kroppen. Att göra detta i labbet är långsamt och dyrt, och även dagens kraftfulla artificiella intelligensverktyg kan ha svårt när endast ett fåtal mätningar finns för ett nytt sjukdomsprotein. I den här artikeln presenteras AdaMBind, ett inlärningssystem utformat för att göra tillförlitliga gissningar om hur starkt läkemedel binder till tidigare osedda mål, även när data är knappa. 
Varför läkemedels–måls "stickighet" spelar roll
När ett läkemedel fungerar är det vanligtvis för att det hakar fast vid ett specifikt protein och förändrar vad det proteinet gör. Styrkan i detta grepp, kallad affinitet, är en nyckelfaktor för att omvandla en lovande molekyl till en verklig behandling. Klassiska labbtekniker kan mäta affinitet mycket exakt, men de kräver specialiserad utrustning, erfaren hantering och mycket tid. Datorbaserade modeller lovar mycket snabbare screening, men de flesta nuvarande djupinlärningsmetoder förutsätter att de ser gott om exempel för varje protein. I verklig läkemedelsupptäckt har många intressanta proteiner bara ett fåtal kända föreningar, så modeller som tränas på vanligt sätt tenderar att överanpassa sig till välstuderade mål och misslyckas på nya.
Lära sig att lära från många små problem
AdaMBind angriper denna utmaning med meta-inlärning, ibland kallad "lära sig att lära." Istället för att behandla hela datamängden som ett stort problem delar metoden upp den i många mindre uppgifter, var och en centrerad kring ett enda protein och alla läkemedel som testats mot det. Modellen tränas sedan över dessa uppgifter så att den skaffar sig en intern utgångspunkt som snabbt kan justeras för ett nytt protein med endast ett fåtal kända mätningar. Under huven representerar systemet läkemedel som grafer av atomer och bindningar, och proteiner som aminosyrasekvenser. Separata neurala nätverk bearbetar respektive sida och kombinerar sedan sina egenskaper för att förutsäga bindningsstyrka, men den avgörande vändningen är hur modellen tränas över uppgifterna.
Från lätta lektioner till svåra
Inte varje uppgift är lika informativ. Vissa protein–läkemedelsuppsättningar är brusiga eller ovanligt svåra och kan vilseleda träningsprocessen om de behandlas på samma sätt som renare uppgifter. AdaMBind lägger till en adaptiv uppgiftsmodul som ständigt poängsätter uppgifterna efter hur väl inlärning på en liten "support"-del överförs till en held-out "query"-del. Uppgifter som ger lägre fel och där inlärningsriktningarna överensstämmer mellan support- och query-seten behandlas som "lättare" och mer pålitliga. Modulen ger dessa uppgifter högre sannolikhetsvikt vid provtagning, så modellen först konsoliderar det den kan lära sig med förtroende och därefter gradvis införlivar svårare uppgifter. Denna lätt-till-svårt-schemaläggning efterliknar hur människor ofta lär sig och gör det slutliga systemet mer stabilt och mindre känsligt för avvikare. 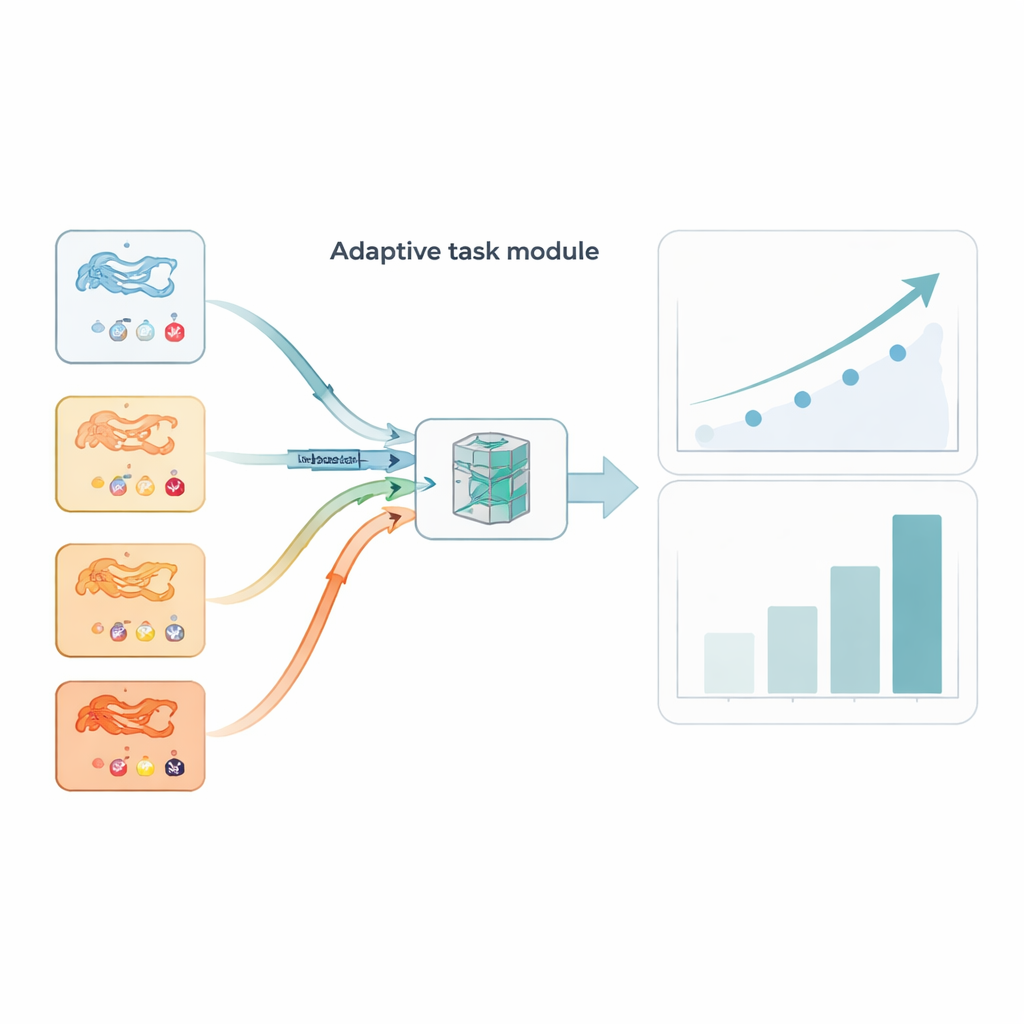
Utstickande i data-knappa förhållanden
Författarna testade AdaMBind på tre standarddatabaser för läkemedels–målsaffinitet—BindingDB, KIBA och Davis—med både generösa och mycket små provstorlekar per protein, och med antingen slumpmässiga uppdelningar eller avsiktligt olikartade testmål. I nästan alla förhållanden slog AdaMBind åtta starka jämförande metoder, särskilt när endast fem kända läkemedels–proteininpar fanns tillgängliga för anpassning till ett nytt mål. Ytterligare tester visade att dess prestanda förblev stark även när det nya proteinet hade få nära släktingar i träningssetet, vilket antyder att modellen inte bara memorerar liknande uppgifter utan extraherar bredare användbara mönster. En strategi med etikettbrus, som lätt perturbterar affinitetsvärden under träningen, förbättrar vidare robustheten genom att avskräcka modellen från att fästa sig för hårt vid möjligen ofullständiga mätningar.
Från benchmark till verkliga läkemedelsledtrådar
För att bedöma dess praktiska värde bad teamet AdaMBind hjälpa till med virtuella screeningproblem som liknar verkliga projekt. På en utmanande dataset där endast en mycket liten andel föreningar verkligen är aktiva mot mål som ESR och TP53 lyckades metoden placera många av de verkliga träffarna högt i rankningen och överträffade andra modeller på mått som premierar "tidig berikning." De tillämpade sedan AdaMBind på det leukemirelaterade proteinet FLT3 och skannade en stor läkemedelsdatabas efter starka bindare. Bland dess toppförslag fanns föreningen staurosporin. Uppföljande dockningssimulationer och laboratoriekinasanalyser bekräftade att staurosporin hämmar både normala och muterade former av FLT3 med sub-nanomolär potens, ännu starkare än en känd klinisk hämmare, vilket visar att modellens förutsägelser kan peka på verkligt kraftfulla molekyler.
En smartare utgångspunkt för framtida läkemedelsjägare
I vardagstermer ger AdaMBind ett sätt för ett AI-system att lära sig goda "instinkter" om läkemedels–proteinsbindning från många små, ofullkomliga lektioner och sedan snabbt tillämpa dessa instinkter när det ställs inför ett nytt, dåligt studerat mål. Genom att avgöra vilka träningsuppgifter som är värda att lita på först och genom att vara relativt okänslig för hur likt ett nytt protein är tidigare exempel, erbjuder metoden en mer pålitlig vägledning för virtuell screening under snäva databudgetar. Det finns förstås utrymme att växa—såsom att införliva rikare 3D-information och att sträva mot verkliga noll-dataförutsägelser—men denna ram utgör ett steg mot snabbare, mer flexibla och mer dataeffektiva upptäckter av framtida läkemedel.
Citering: Wan, M., Zhao, Y., Zhang, Y. et al. A meta learning and task adaptive approach for drug target affinity prediction. Nat Commun 17, 3734 (2026). https://doi.org/10.1038/s41467-026-70554-5
Nyckelord: förutsägelse av läkemedels–målsaffinitet, meta-inlärning, virtuell screening, few-shot-inlärning, FLT3-hämmare