Clear Sky Science · it
Un approccio meta-apprendimento e adattivo al compito per la predizione dell’affinità target-farmaco
Insegnare ai computer a scegliere medicine migliori
Scoprire nuovi farmaci spesso significa testare milioni di molecole possibili per vedere quali si legano ai giusti target proteici nell’organismo. Farlo in laboratorio è lento e costoso, e anche gli odierni potenti strumenti di intelligenza artificiale possono inciampare quando sono disponibili solo poche misurazioni per una nuova proteina legata a una malattia. Questo articolo presenta AdaMBind, un sistema di apprendimento progettato per fare previsioni affidabili su quanto fortemente i farmaci si legheranno a target mai visti prima, anche quando i dati scarseggiano. 
Perché la “adesione” farmaco–target è importante
Quando un farmaco funziona, di solito è perché si aggancia a una proteina specifica e ne altera la funzione. La forza di questa presa, chiamata affinità, è un elemento chiave per trasformare una molecola promettente in una terapia reale. Le tecniche di laboratorio classiche possono misurare l’affinità con grande precisione, ma richiedono strumenti specializzati, operatori esperti e molto tempo. I modelli computazionali promettono di accelerare enormemente lo screening, ma la maggior parte degli approcci di deep learning attuali presuppone di disporre di molti esempi per ogni proteina. Nella realtà della scoperta di farmaci, molte proteine interessanti hanno solo pochi composti noti, quindi i modelli addestrati nel modo tradizionale tendono a sovradattarsi ai target meglio studiati e a fallire sui nuovi.
Imparare a imparare da molti piccoli problemi
AdaMBind affronta questa sfida con il meta-apprendimento, a volte chiamato “imparare a imparare”. Invece di trattare l’intero dataset come un unico grande problema, il metodo lo suddivide in molti compiti più piccoli, ciascuno incentrato su una singola proteina e su tutti i farmaci testati contro di essa. Il modello si allena attraverso questi compiti in modo da acquisire un punto di partenza interno che può essere rapidamente adattato a una nuova proteina usando solo poche misurazioni note. Sotto il cofano, il sistema rappresenta i farmaci come grafi di atomi e legami, e le proteine come sequenze di amminoacidi. Reti neurali separate elaborano ciascuna componente e poi combinano le caratteristiche per prevedere la forza del legame, ma la svolta cruciale sta in come il modello viene addestrato attraverso i compiti.
Dalle lezioni facili a quelle difficili
Non tutti i compiti sono ugualmente informativi. Alcune raccolte proteina–farmaco sono rumorose o insolitamente difficili e possono fuorviare il processo di addestramento se trattate allo stesso modo dei compiti più puliti. AdaMBind aggiunge un modulo di compito adattivo che valuta costantemente i compiti in base a quanto bene l’apprendimento su un piccolo sottoinsieme di “supporto” si trasferisce a un sottoinsieme di “query” tenuto da parte. I compiti che producono errori minori e i cui vettori di apprendimento concordano tra insiemi di supporto e query sono considerati “più facili” e più affidabili. Il modulo assegna a questi compiti un peso di campionamento maggiore, così il modello consolida prima ciò che può imparare con fiducia e poi incorpora gradualmente i compiti più difficili. Questa programmazione da facile a difficile imita il modo in cui le persone spesso apprendono e rende il sistema finale più stabile e meno sensibile agli outlier. 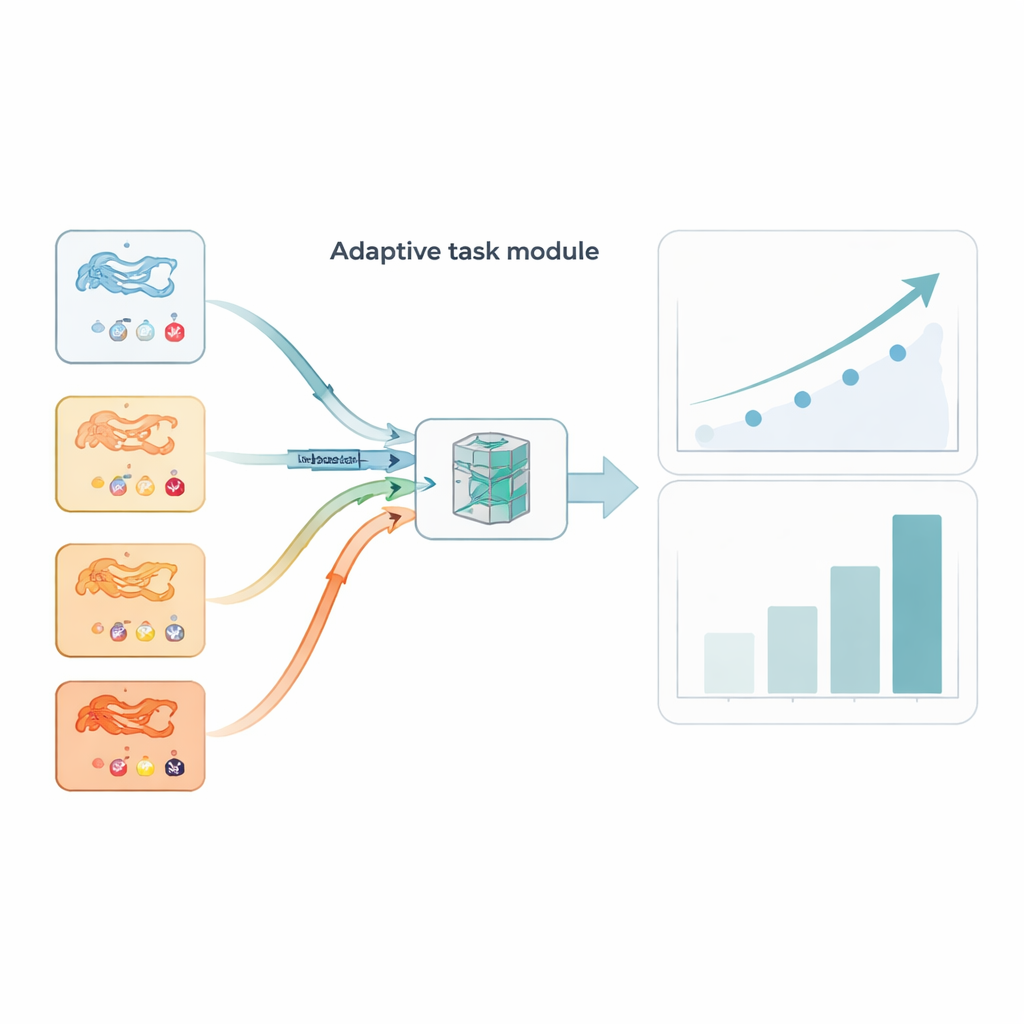
Distinguersi in condizioni di scarsezza di dati
Gli autori hanno testato AdaMBind su tre collezioni standard di affinità farmaco–target—BindingDB, KIBA e Davis—utilizzando sia campioni numerosi sia dimensioni molto ridotte per proteina, e con divisioni casuali o con target di test intenzionalmente dissimili. In quasi tutte le condizioni, AdaMBind ha superato otto metodi di confronto robusti, soprattutto quando erano disponibili solo cinque coppie farmaco–proteina note per adattarsi a un nuovo target. Ulteriori test hanno mostrato che le sue prestazioni rimanevano solide anche quando la nuova proteina aveva pochi parenti stretti nel set di addestramento, suggerendo che il modello non sta semplicemente memorizzando compiti simili ma estraendo schemi utili in senso più ampio. Una strategia di rumore sulle etichette, che altera lievemente i valori di affinità durante l’addestramento, migliora ulteriormente la robustezza evitando che il modello si aggrappi troppo a misurazioni potenzialmente imperfette.
Dai benchmark a potenziali candidati farmaceutici reali
Per valutarne il valore pratico, il team ha impiegato AdaMBind in problemi di screening virtuale che assomigliano a progetti del mondo reale. Su un dataset impegnativo in cui solo una piccolissima frazione di composti è veramente attiva contro target come ESR e TP53, il metodo è stato in grado di posizionare molti dei veri hit nella parte alta della lista di ranking, superando altri modelli su misure che premiano il “arricchimento precoce”. Hanno poi applicato AdaMBind alla proteina legata alla leucemia FLT3, esaminando un ampio database di composti per identificare forti binder. Tra le principali proposte è comparso il composto staurosporina. Simulazioni di docking successive e saggi enzimatici di laboratorio hanno confermato che la staurosporina inibisce sia la forma normale sia quella mutata di FLT3 con potenza sub-nanomolare, anche più efficacemente di un noto inibitore clinico, dimostrando che le previsioni del modello possono indicare molecole realmente potenti.
Un punto di partenza più intelligente per i futuri cacciatori di farmaci
In termini pratici, AdaMBind offre un modo per un sistema di IA di apprendere buoni “istinti” sul legame farmaco–proteina da molte piccole e imperfette lezioni, per poi applicare rapidamente quegli istinti di fronte a un nuovo target poco studiato. Decidendo quali compiti di addestramento fidarsi prima e rimanendo relativamente insensibile a quanto un nuovo target somigli agli esempi passati, il metodo offre una guida più affidabile per lo screening virtuale con budget di dati limitati. Pur essendoci margine di miglioramento—per esempio integrando informazioni 3D più ricche e spingendo verso vere predizioni con zero dati—questa struttura rappresenta un passo verso una scoperta di farmaci più rapida, più flessibile e più efficiente in termini di dati.
Citazione: Wan, M., Zhao, Y., Zhang, Y. et al. A meta learning and task adaptive approach for drug target affinity prediction. Nat Commun 17, 3734 (2026). https://doi.org/10.1038/s41467-026-70554-5
Parole chiave: predizione affinità farmaco-target, meta-apprendimento, screening virtuale, few-shot learning, inibitori FLT3