Clear Sky Science · de
Ein meta‑lernender und aufgabenadaptiver Ansatz zur Vorhersage der Arzneimittel‑Ziel‑Affinität
Computern beibringen, bessere Medikamente auszuwählen
Neue Medikamente zu finden bedeutet oft, Millionen möglicher Moleküle daraufhin zu testen, welche an die richtigen Proteinziele im Körper binden. Experimente im Labor sind langsam und teuer, und selbst heutige leistungsfähige KI‑Werkzeuge geraten ins Straucheln, wenn für ein neues Krankheitseiweiß nur wenige Messwerte vorliegen. Dieses Papier stellt AdaMBind vor, ein Lernsystem, das zuverlässige Schätzungen zur Bindungsstärke von Wirkstoffen an bislang unbekannte Ziele liefern soll — selbst wenn nur wenig Daten verfügbar sind. 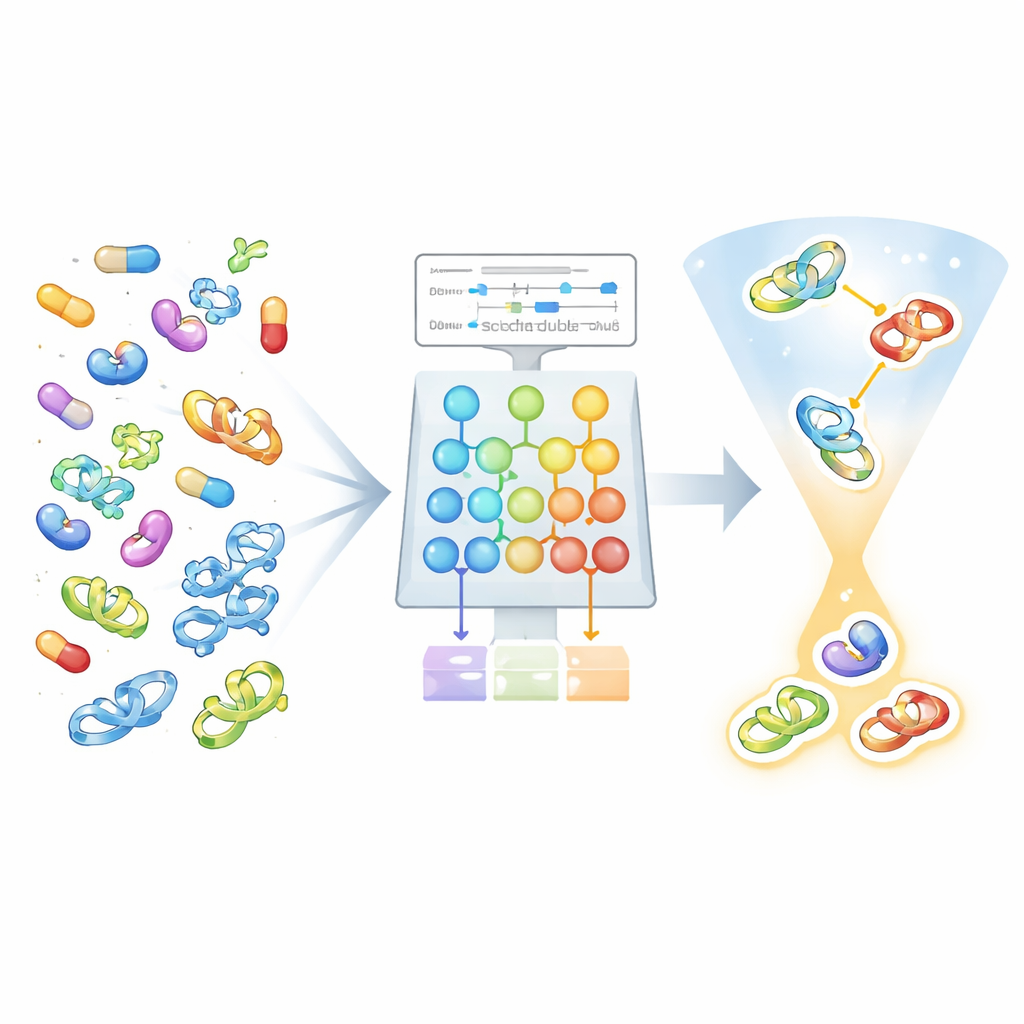
Warum die Bindungsstärke zwischen Wirkstoff und Ziel wichtig ist
Wenn ein Wirkstoff wirkt, liegt das meist daran, dass er an ein bestimmtes Protein bindet und dessen Funktion verändert. Die Stärke dieses Griffs, genannt Affinität, ist ein entscheidender Faktor, um ein vielversprechendes Molekül in eine echte Therapie zu verwandeln. Klassische Labormethoden messen Affinitäten sehr genau, benötigen jedoch spezialisierte Geräte, erfahrene Bedienung und viel Zeit. Computermodelle versprechen deutlich schnelleres Screening, doch die meisten aktuellen Deep‑Learning‑Ansätze gehen davon aus, für jedes Protein viele Beispiele zu sehen. In der realen Wirkstoffforschung haben viele interessante Proteine dagegen nur wenige bekannte Verbindungen, sodass klassisch trainierte Modelle dazu neigen, sich an gut untersuchten Zielen festzubeißen und bei neuen zu versagen.
Aus vielen kleinen Problemen lernen, wie man lernt
AdaMBind begegnet dieser Herausforderung mit Meta‑Lernen, manchmal „Lernen zu lernen“ genannt. Statt den gesamten Datensatz als ein großes Problem zu behandeln, teilt die Methode ihn in viele kleinere Aufgaben auf, die jeweils ein einzelnes Protein und alle gegen dieses getesteten Wirkstoffe umfassen. Das Modell trainiert über diese Aufgaben hinweg, sodass es einen internen Startpunkt erwirbt, der sich mit nur wenigen bekannten Messwerten schnell an ein neues Protein anpassen lässt. Unter der Haube werden Wirkstoffe als Graphen aus Atomen und Bindungen und Proteine als Aminosäuresequenzen dargestellt. Getrennte neuronale Netze verarbeiten jede Seite und kombinieren dann ihre Merkmale zur Vorhersage der Bindungsstärke — der entscheidende Dreh ist jedoch, wie das Modell über die Aufgaben hinweg trainiert wird.
Von einfachen Lektionen zu schwierigen
Nicht jede Aufgabe liefert gleich viel Information. Manche Protein‑Wirkstoff‑Sammlungen sind laut oder ungewöhnlich schwierig und können den Trainingsprozess in die Irre führen, wenn man sie gleichbehandelt wie sauberere Aufgaben. AdaMBind ergänzt ein adaptives Aufgabenmodul, das Aufgaben kontinuierlich danach bewertet, wie gut Lernen auf einer kleinen „Support“‑Teilmenge auf eine zurückgehaltene „Query“‑Teilmenge übertragbar ist. Aufgaben, die geringere Fehler liefern und bei denen die Lernrichtungen zwischen Support‑ und Query‑Sets übereinstimmen, gelten als „einfacher“ und zuverlässiger. Das Modul gibt diesen Aufgaben ein höheres Sampling‑Gewicht, sodass das Modell zunächst das konsolidiert, was es mit Vertrauen lernen kann, und dann schrittweise schwierigere Aufgaben einbezieht. Dieses Easy‑to‑Hard‑Scheduling ahmt nach, wie Menschen oft lernen, und macht das Endsystem stabiler und weniger anfällig für Ausreißer. 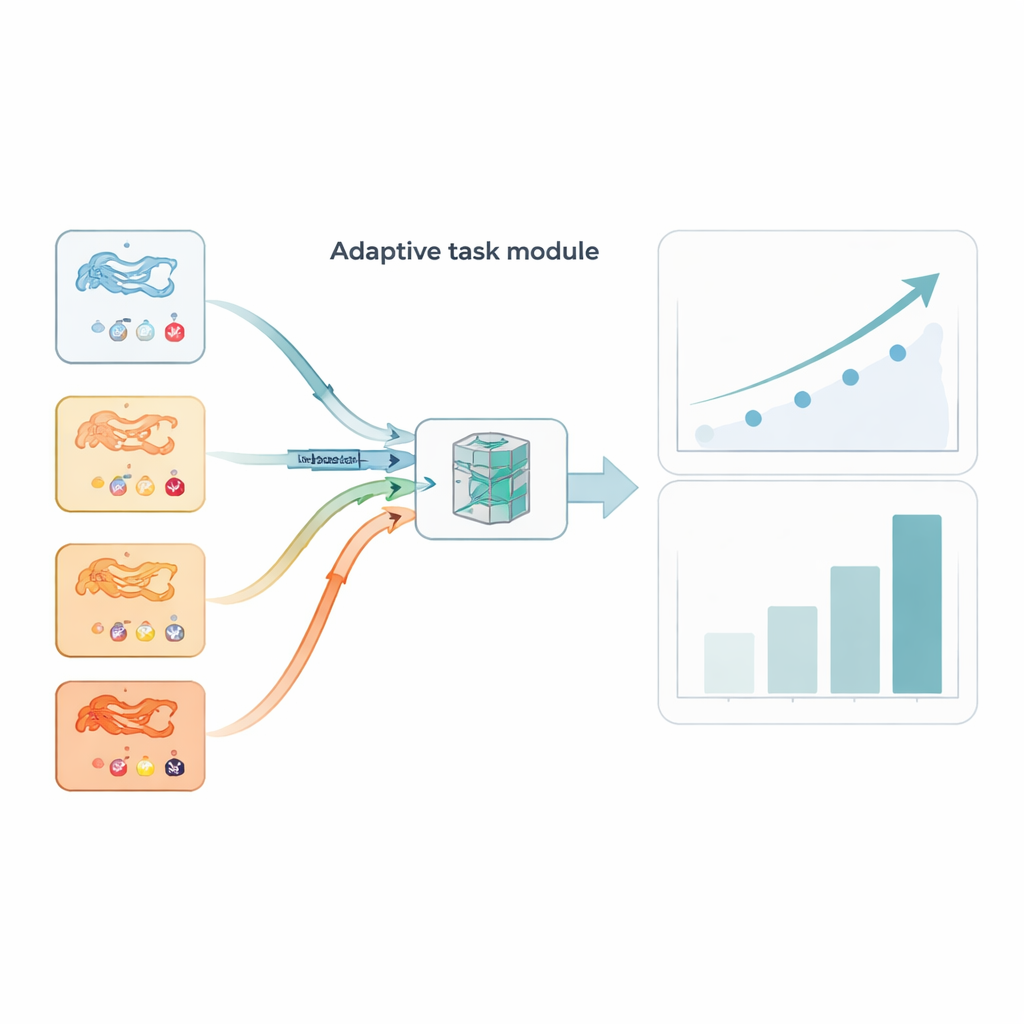
Hervorragende Leistung unter datenarmen Bedingungen
Die Autoren testeten AdaMBind an drei etablierten Datensätzen für Arzneimittel‑Ziel‑Affinitäten — BindingDB, KIBA und Davis — sowohl mit großzügigen als auch sehr kleinen Stichprobengrößen pro Protein und mit zufälligen Aufteilungen oder absichtlich unähnlichen Testzielen. In nahezu allen Szenarien übertraf AdaMBind acht starke Vergleichsmethoden, besonders wenn nur fünf bekannte Wirkstoff‑Protein‑Paare zur Anpassung an ein neues Ziel verfügbar waren. Weitere Tests zeigten, dass die Leistung auch dann robust blieb, wenn das neue Protein nur wenige nahe Verwandte im Trainingssatz hatte, was nahelegt, dass das Modell nicht nur ähnliche Aufgaben auswendig lernt, sondern allgemein nützliche Muster extrahiert. Eine Label‑Noise‑Strategie, die die Affinitätswerte während des Trainings leicht stört, verbessert die Robustheit zusätzlich, indem sie das Modell davon abhält, sich zu sehr an möglicherweise fehlerhafte Messwerte zu klammern.
Von Benchmarks zu realen Wirkstoffkandidaten
Um den praktischen Wert zu prüfen, setzte das Team AdaMBind für virtuelle Screening‑Aufgaben ein, die realen Projekten ähneln. In einem herausfordernden Datensatz, in dem nur ein winziger Bruchteil der Verbindungen gegen Ziele wie ESR und TP53 wirklich aktiv ist, schaffte es die Methode, viele der echten Treffer weit oben in der Rangliste zu platzieren und übertraf andere Modelle bei Metriken, die eine frühe Anreicherung belohnen. Anschließend wandten sie AdaMBind auf das leukämieassoziierte Protein FLT3 an und durchsuchten eine große Wirkstoffdatenbank nach starken Bindern. Unter den Top‑Vorschlägen fand sich das Molekül Staurosporine. Nachfolgende Docking‑Simulationen und Laborkinase‑Assays bestätigten, dass Staurosporine sowohl die normale als auch die mutierte Form von FLT3 mit sub‑nanomolarer Potenz hemmt — sogar stärker als ein bekannter klinischer Inhibitor — und zeigen, dass die Vorhersagen des Modells auf wirklich potente Moleküle hinweisen können.
Ein klügerer Ausgangspunkt für künftige Wirkstoffforscher
Vereinfacht gesagt bietet AdaMBind einer KI‑Anwendung die Möglichkeit, aus vielen kleinen, unvollkommenen Lektionen gute „Instinkte“ für Wirkstoff‑Protein‑Bindung zu erlernen und diese Instinkte schnell auf ein neues, wenig untersuchtes Ziel anzuwenden. Indem das System entscheidet, welchen Trainingsaufgaben es zuerst vertraut, und relativ unempfindlich gegenüber der Ähnlichkeit eines neuen Proteins zu früheren Beispielen bleibt, liefert der Ansatz eine verlässlichere Orientierung für virtuelles Screening unter knappen Datenbedingungen. Zwar gibt es noch Raum für Verbesserungen — etwa die Einbindung reichhaltigerer 3D‑Informationen oder das Vorstoßen zu echten Null‑Daten‑Vorhersagen — doch dieses Rahmenwerk ist ein Schritt hin zu schnellerer, flexiblerer und daten‑effizienterer Entdeckung künftiger Medikamente.
Zitation: Wan, M., Zhao, Y., Zhang, Y. et al. A meta learning and task adaptive approach for drug target affinity prediction. Nat Commun 17, 3734 (2026). https://doi.org/10.1038/s41467-026-70554-5
Schlüsselwörter: Vorhersage der Arzneimittel‑Ziel‑Affinität, Meta‑Lernen, virtuelles Screening, Few‑Shot‑Lernen, FLT3‑Inhibitoren