Clear Sky Science · sv
Uppklarning av rollen för redoxaktiva platser i kvävedopat ceriumoxid för associativ ammoniaknedbrytning
Att förvandla en välkänd kemikalie till ren bränsle
Ammoniak är mest känd som ett stickande rengöringsmedel och en viktig ingrediens i gödningsmedel, men den bär också på en dold förmåga: den kan lagra väte, den lätta gas som många hoppas ska driva en lågkolkframtid. För att utnyttja den potentialen måste vi effektivt utvinna väte ur ammoniak utan att producera koldioxid, med katalysatorer som fungerar bra vid relativt låga temperaturer. Denna studie visar hur subtila förändringar på ytan av ett fast material kan låsa upp ett nytt sätt att slå sönder ammoniak, kringgå långvariga begränsningar i katalysatordesign och peka mot renare, mer effektiv väteproduktion.
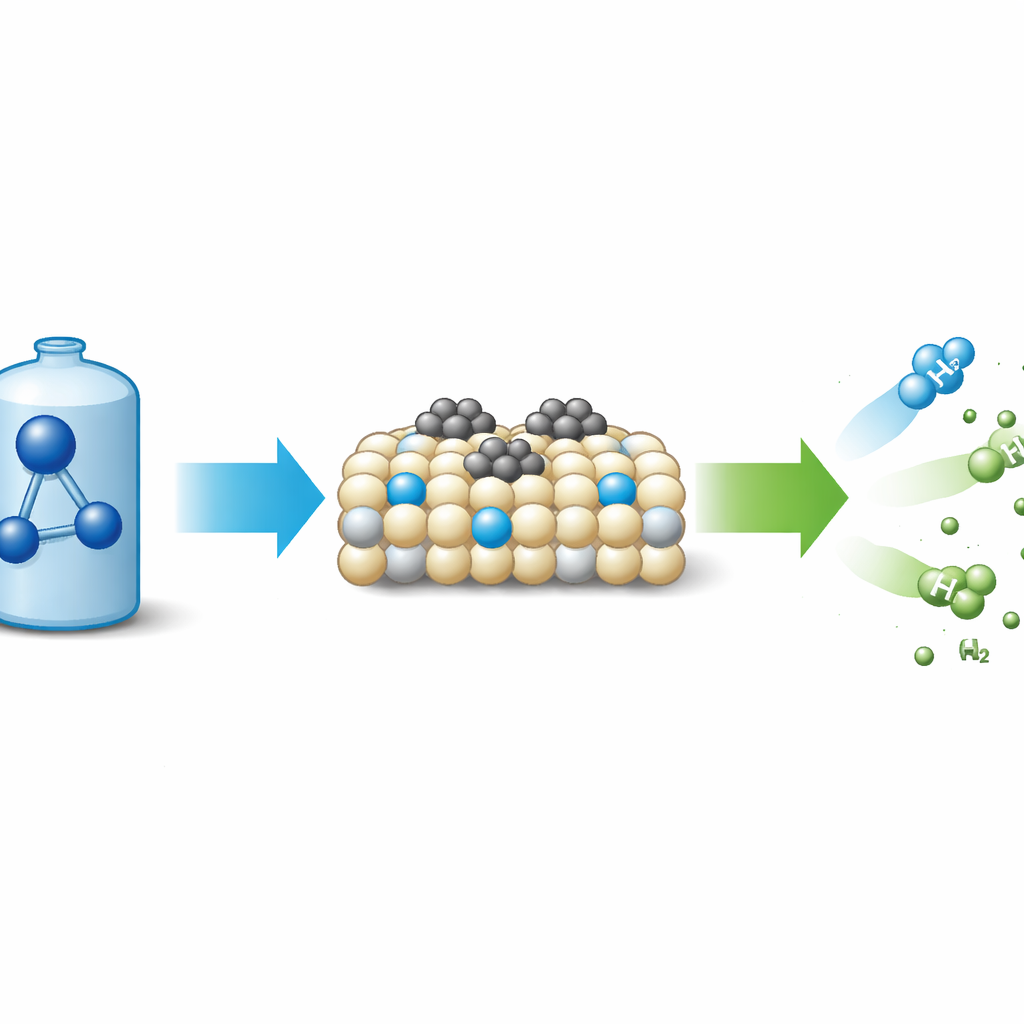
Varför ammoniak betyder något för grön energi
Ammoniak (NH₃) rymmer mer väte per liter än komprimerad vätgas och är lätt att förflytta som vätska. När den sönderdelas ger den väte och kväve—inga koldioxidutsläpp. Utmaningen är att denna reaktion kräver mycket energi, så industrin förlitar sig på kraftfulla katalysatorer för att sänka temperatur och kostnad. Rutenium, en ädelmetall, är för närvarande standarden för att påskynda ammoniakbrytning, men den följer ett välkänt mönster: först bryts N–H‑bindningarna på metallytan, sedan parar sig kväveatomerna och lämnar som N₂. Denna ”dissociativa” väg är fångad av en grundläggande avvägning: metaller som binder kväve starkt hjälper till att bryta N–H‑bindningar men håller sedan kvar kväveatomerna för hårt, vilket gör det svårt för N₂ att desorbsera. Denna avvägning, fångad i ett samband känt som Brønsted–Evans–Polanyi‑skalning, har blivit en viktig flaskhals för bättre katalysatorer.
Låna ett knep från naturen
Naturens kvävebearbetande enzymer, kallade nitrogenaser, undviker denna fälla genom att följa ett annat manus. Istället för att riva N≡N‑bindningar i ett enda steg tillsätter och tar de bort väte stegvis vid speciella redoxaktiva platser, och bildar och omformar N–H och N–N‑bindningar på ett mer kooperativt sätt. Inspirerade av detta frågade författarna om en fast katalysator kunde efterlikna ett sådant ”associativt” beteende, där ammoniak interagerar med ett reaktivt centrum som både kan ge och ta elektroner och tillfälligt lagra kväve. De fokuserade på ceriumoxid (CeO₂), ett välkänt ”syretransportör”-material vars atomer lätt kan skifta mellan oxidationstillstånd. Genom att införa en liten mängd kväve i gitterstrukturen—skapa kvävedopat ceriumoxid—avsiktade de att göra vissa gitterplatser till redoxaktiva kvävecentrum som kanske kunde delta direkt i att bryta och bilda kvävbindningar.
Design av en tvåplatserad katalysator
Teamet syntetiserade ceriumoxid med en mild gelmetod och behandlade den sedan med strömmande ammoniak vid olika temperaturer för att introducera kväveatomer i kristallen. Dessa kvävedopanter skapade extra defekter och syrevakanser, gjorde ytan lättare att reducera och ökade dess elektronavgivande förmåga. När små ruteniumnanopartiklar deponerades på detta kvävedopade stöd, bröt den resulterande katalysatorn (Ru/N‑CeO₂) ned ammoniak mycket effektivare än samma mängd rutenium på odopat ceriumoxid. Vid 450–500 °C nådde det optimerade materialet nästan fullständig konversion med lägre metallbelastning än många toppsystem och förblev stabilt i minst 70 timmar. Mätningar av reaktionshastigheter visade att den uppenbara aktiveringsenergin sjönk långt under det typiska intervallet för ruteniumkatalysatorer, ett starkt tecken på att reaktionsvägen själv hade förändrats.
Se en ny väg i arbete
För att ta reda på vad som verkligen hände på ytan kombinerade forskarna infrarödspektroskopi under operativa förhållanden, isotopmärkningsexperiment och avancerade datorsimuleringar. Infraröda signaler visade uppkomsten av kväve–kväve och N–N–H‑typiska arter på det kvävedopade stödet, vilket tyder på att inkommande ammoniakmolekyler bildade bindningar direkt med gitterkväveatomer. Noggrant tidsbestämda pulser av ammoniak med tyngre kväve (¹⁵N) visade att en del av kvävet i produkt‑N₂ kom från dessa dopade gitterplatser, inte bara från gasen. Massspektrometridata gjorde det möjligt för teamet att kvantifiera bidragen från två samtidiga vägar: en konventionell dissociativ väg på rutenium och en associativ, så kallad Mars–van Krevelen‑väg där gitterkvävet tillfälligt lämnar och senare fylls på av kväve från ammoniak. Ungefär en tredjedel av det bildade N₂ följde denna nya associativa väg.
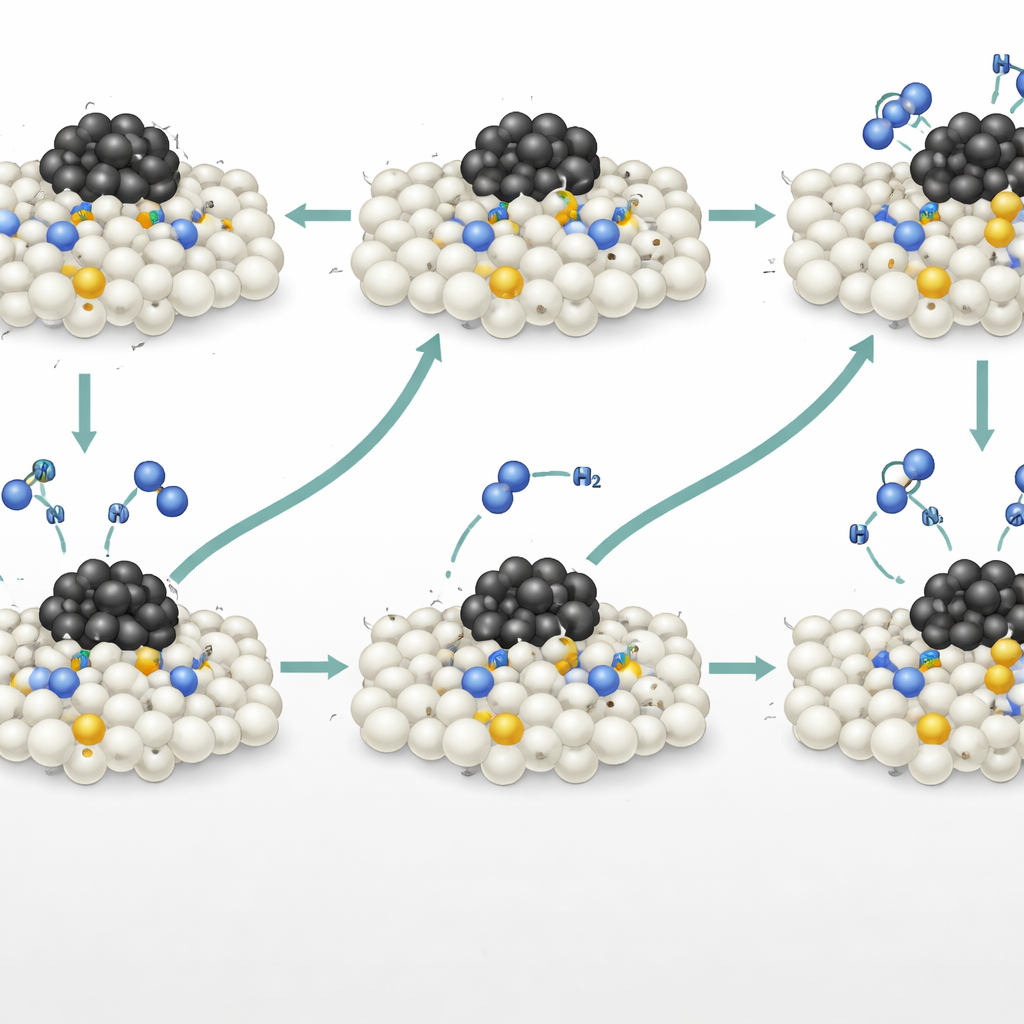
Var de mest aktiva platserna finns
Datormodeller baserade på maskininlärningsaccelererade simuleringar, kontrollerade mot detaljerade kvantmekaniska beräkningar, kartlade energilandskapet för denna associativa väg. De visade att kvävedopanter nära—men inte direkt bundna till—ruteniumpartiklarna erbjöd den lägsta totala energibarriären. Dessa ”proximala” platser känner metallens elektroniska påverkan tillräckligt starkt för att underlätta N–H‑bindningsbrytning, men är tillräckligt långt bort för att kvävemolekyler fortfarande ska kunna avgå utan att fastna vid rutenium. Däremot binder kväve precis vid metal‑stödsgränsen för starkt och saktar ner kvävefrigörelsen, medan avlägsna platser saknar tillräcklig hjälp från metallen. Denna icke‑intuiva upptäckt kullkastar den vanliga föreställningen att metal‑stödsgränsen alltid är den mest aktiva regionen, och lyfter istället fram en idealisk avståndszon där gitterkväve agerar som ett kraftfullt, självständigt aktivt centrum.
En ny spelbok för väte från ammoniak
Genom att bevisa att kvävedopanter i ett redoxaktivt stöd kan fungera som egna katalytiska platser, arbeta sida vid sida med metallnanopartiklar men genom en distinkt mekanism, visar detta arbete en konkret väg att bryta sig loss från långvariga skalningsbegränsningar i ammoniaknedbrytning. Noggrann placering och stabilisering av dessa gitterkväveplatser tillåter att ammoniak bryts ner genom en associativ väg som kopplar N–H‑bindningsklyvning med N–N‑bindningsbildning, vilket sänker energibehovet och möjliggör hög aktivitet vid mildare förhållanden. För icke‑specialister är slutsatsen att katalysatorns ”bakgrundstöd” inte bara är ett inert skelett: när det kemiskt utformas kan det dela arbetsbördan med ädelmetaller och till och med omforma själva reaktionen. Den insikten kan vägleda utformningen av nästa generations katalysatorer för ammoniakbaserade vätessystem och andra renenergireaktioner som idag verkar begränsade av fundamentala avvägningar.
Citering: Ye, D., Luo, M., Liu, X. et al. Unravelling the role of redox active sites in nitrogen doped cerium oxide for associative ammonia decomposition. Nat Commun 17, 3892 (2026). https://doi.org/10.1038/s41467-026-70330-5
Nyckelord: ammoniaknedbrytning, väteproduktion, ruteniumkatalysator, kvävedopat ceria, heterogen katalys