Clear Sky Science · fr
Décrypter le rôle des sites rédox actifs dans l’oxyde de cérium dopé à l’azote pour la décomposition associative de l’ammoniac
Transformer un produit chimique familier en carburant propre
L’ammoniac est surtout connu comme un liquide de nettoyage âcre et comme un ingrédient clé des engrais, mais il possède aussi un talent caché : il peut stocker de l’hydrogène, le gaz léger que beaucoup espèrent voir alimenter un avenir bas‑carbone. Pour exploiter ce potentiel, il faut extraire l’hydrogène de l’ammoniac de façon efficace sans produire de dioxyde de carbone, en utilisant des catalyseurs performants à des températures relativement basses. Cette étude montre comment des modifications subtiles de la surface d’un solide peuvent ouvrir une nouvelle voie pour décomposer l’ammoniac, contournant des limites de longue date dans la conception des catalyseurs et ouvrant la voie à une production d’hydrogène plus propre et plus efficace.
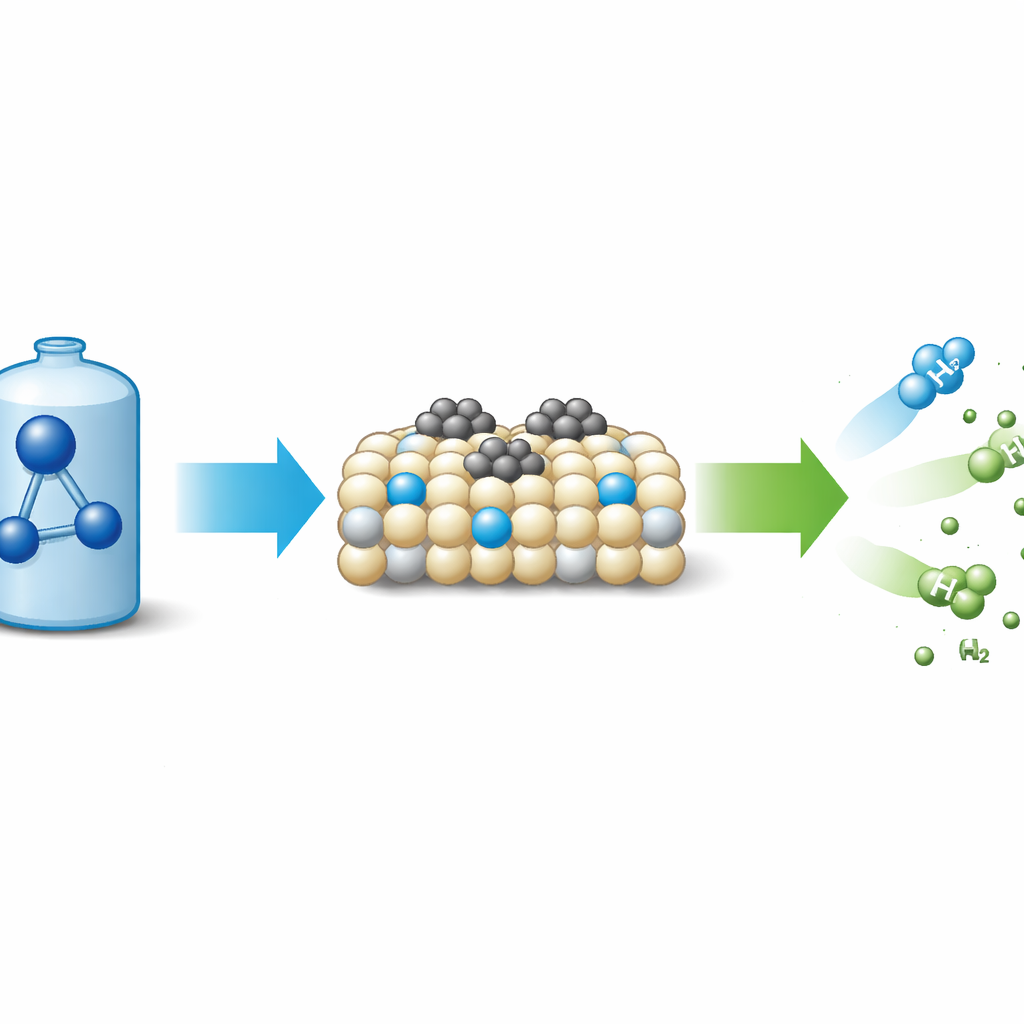
Pourquoi l’ammoniac compte pour l’énergie verte
L’ammoniac (NH₃) contient plus d’hydrogène par litre que l’hydrogène comprimé et se liquéfie et se transporte facilement. Lorsqu’il est décomposé, il produit de l’hydrogène et de l’azote—sans émissions de carbone. Le défi tient au fait que cette réaction demande beaucoup d’énergie, si bien que l’industrie s’appuie sur des catalyseurs puissants pour diminuer la température et le coût. Le ruthénium, métal précieux, est actuellement la référence pour accélérer la décomposition de l’ammoniac, mais il suit un schéma bien connu : d’abord les liaisons N–H se rompent à la surface du métal, puis les atomes d’azote s’apparient et s’échappent sous forme de N₂. Cette voie « dissociative » est prise au piège d’un compromis fondamental : les métaux qui lient fortement l’azote facilitent la rupture des liaisons N–H mais retiennent ensuite trop les atomes d’azote, rendant difficile la désorption du N₂. Ce compromis, décrit par la relation de Brønsted–Evans–Polanyi, est devenu un obstacle majeur à de meilleurs catalyseurs.
Emprunter un tour à la nature
Les enzymes de traitement de l’azote de la nature, appelées nitrogénases, évitent ce piège en suivant un autre scénario. Plutôt que de déchirer la liaison N≡N en une seule étape, elles ajoutent et retirent des hydrogènes pas à pas sur des sites rédox‑actifs spéciaux, formant et réarrangeant les liaisons N–H et N–N de manière plus coopérative. S’inspirant de cela, les auteurs se sont demandé si un catalyseur solide pouvait imiter ce comportement « associatif », où l’ammoniac interagit avec un site réactif capable à la fois de donner et de recevoir des électrons et de stocker temporairement de l’azote. Ils se sont concentrés sur l’oxyde de cérium (CeO₂), matériau bien connu pour son rôle de « navette à oxygène » dont les atomes peuvent facilement changer d’état d’oxydation. En insérant une petite quantité d’azote dans le réseau—créant un oxyde de cérium dopé à l’azote—ils visaient à transformer certains sites du réseau en centres azotés rédox‑actifs susceptibles de participer directement à la rupture et à la formation des liaisons azote.
Concevoir un catalyseur à double site
L’équipe a synthétisé l’oxyde de cérium par une voie sol‑gel douce puis l’a traité avec un flux d’ammoniac à différentes températures pour introduire des atomes d’azote dans le cristal. Ces dopants azotés ont généré des défauts supplémentaires et des vacants d’oxygène, rendant la surface plus facilement réductible et renforçant sa capacité à donner des électrons. Lorsque de minuscules nanoparticules de ruthénium ont été déposées sur ce support dopé à l’azote, le catalyseur obtenu (Ru/N‑CeO₂) a décomposé l’ammoniac bien plus efficacement que la même quantité de ruthénium sur de l’oxyde de cérium non dopé. À 450–500 °C, le matériau optimisé atteignait une conversion quasi complète avec une charge métallique plus faible que de nombreux systèmes de pointe et restait stable pendant au moins 70 heures. Les mesures des vitesses de réaction ont montré que l’énergie d’activation apparente chutait bien en dessous de la plage typique des catalyseurs au ruthénium, indice fort que le mécanisme réactionnel lui‑même avait changé.
Observer une nouvelle voie en action
Pour découvrir ce qui se passait réellement à la surface, les chercheurs ont combiné la spectroscopie infrarouge en conditions opératoires, des expériences de marquage isotopique et des simulations informatiques avancées. Les signaux infrarouges ont révélé l’apparition d’espèces azote‑azote et de types N–N–H sur le support dopé à l’azote, suggérant que des molécules d’ammoniac entrantes formaient des liaisons directement avec des atomes d’azote du réseau. Des impulsions soigneusement chronométrées d’ammoniac contenant de l’azote plus lourd (¹⁵N) ont montré qu’une partie de l’azote dans le N₂ produit provenait de ces sites du réseau dopés, et pas seulement du gaz. Les données de spectrométrie de masse ont permis à l’équipe de quantifier les contributions de deux voies simultanées : une voie dissociative conventionnelle sur le ruthénium et une voie associative dite Mars–van‑Krevelen dans laquelle l’azote du réseau quitte temporairement le site puis est ensuite remplacé par de l’azote provenant de l’ammoniac. Environ un tiers du N₂ formé suivait cette nouvelle voie associative.
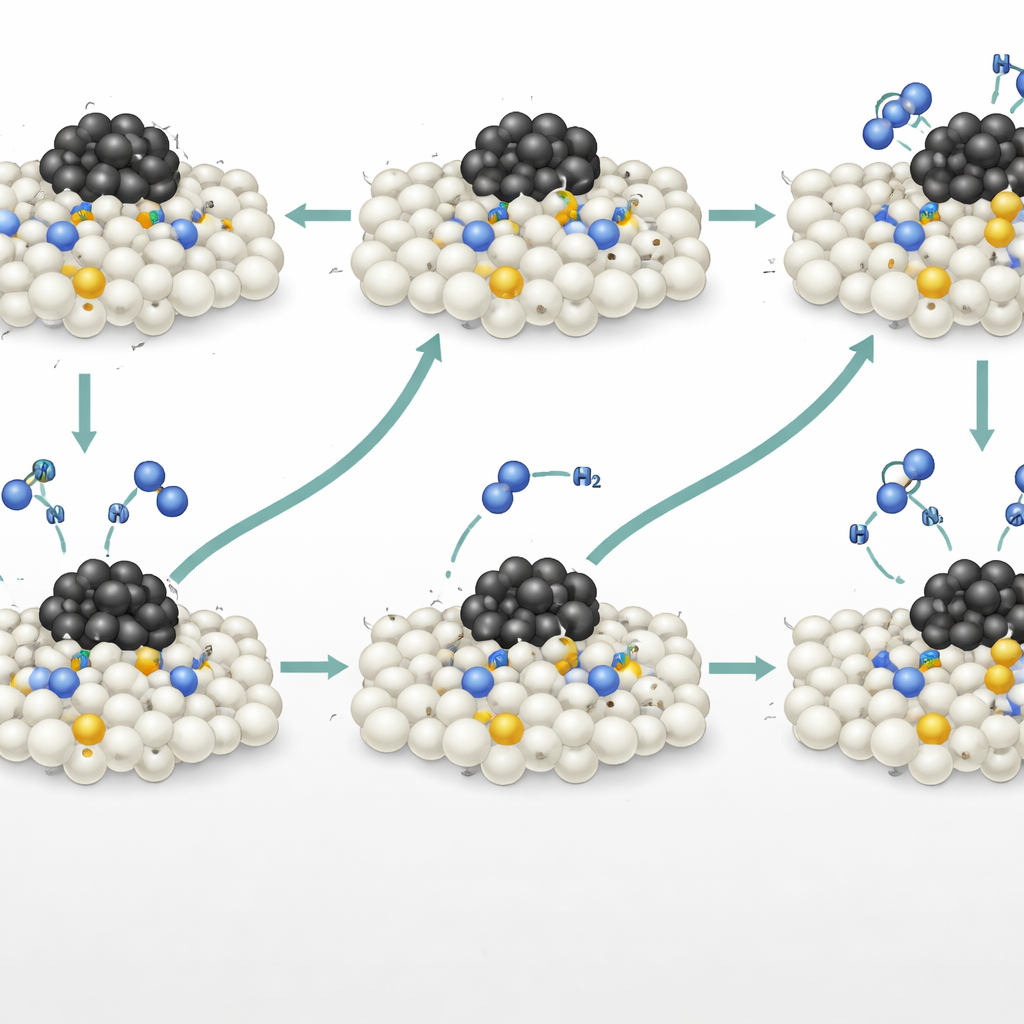
Où se trouvent les sites les plus actifs
Des modèles informatiques basés sur des simulations accélérées par apprentissage automatique, vérifiés par des calculs quantiques détaillés, ont cartographié le paysage énergétique de cette voie associative. Ils ont montré que les dopants azotés situés à proximité—mais pas directement liés—aux particules de ruthénium offraient la barrière énergétique globale la plus basse. Ces sites « proximaux » subissent l’influence électronique du métal suffisamment pour faciliter la rupture des liaisons N–H, tout en étant assez éloignés pour que les molécules d’azote puissent partir sans rester collées au ruthénium. En revanche, l’azote situé exactement à la frontière métal‑support se lie trop fortement et ralentit la libération de l’azote, tandis que les sites plus éloignés ne reçoivent pas suffisamment d’aide du métal. Cette conclusion contre‑intuitive remet en cause la croyance courante selon laquelle l’interface métal‑support est toujours la région la plus active, et met en lumière une « zone optimale » où l’azote du réseau agit comme un centre actif puissant et indépendant.
Un nouveau mode d’emploi pour produire de l’hydrogène à partir de l’ammoniac
En démontrant que des dopants azotés dans un support rédox‑actif peuvent agir comme des sites catalytiques à part entière, travaillant aux côtés de nanoparticules métalliques mais par un mécanisme distinct, ce travail montre une voie concrète pour se libérer des limites d’échelle établies de longue date dans la décomposition de l’ammoniac. Positionner et stabiliser soigneusement ces sites azotés du réseau permet de décomposer l’ammoniac via une voie associative qui couple la rupture des liaisons N–H à la formation des liaisons N–N, réduisant les besoins énergétiques et permettant une activité élevée dans des conditions plus douces. Pour le non‑spécialiste, la conclusion est que le support « de fond » d’un catalyseur n’est pas simplement un échafaudage inerte : lorsqu’il est conçu chimiquement, il peut partager la charge avec les métaux précieux et même remodeler la réaction elle‑même. Cette idée pourrait guider la conception de catalyseurs de nouvelle génération pour des systèmes d’hydrogène à base d’ammoniac et d’autres réactions pour l’énergie propre qui semblent aujourd’hui limitées par des compromis fondamentaux.
Citation: Ye, D., Luo, M., Liu, X. et al. Unravelling the role of redox active sites in nitrogen doped cerium oxide for associative ammonia decomposition. Nat Commun 17, 3892 (2026). https://doi.org/10.1038/s41467-026-70330-5
Mots-clés: décomposition de l’ammoniac, production d’hydrogène, catalyseur au ruthénium, cérium dopé à l’azote, catalyse hétérogène