Clear Sky Science · en
Unravelling the role of redox active sites in nitrogen doped cerium oxide for associative ammonia decomposition
Turning a Familiar Chemical into Clean Fuel
Ammonia is best known as a pungent cleaning fluid and a key ingredient in fertilizers, but it also carries a hidden talent: it can store hydrogen, the light gas many hope will power a low‑carbon future. To tap that potential, we must pull hydrogen efficiently out of ammonia without producing carbon dioxide, using catalysts that work well at relatively low temperatures. This study shows how subtle changes to the surface of a solid material can unlock a new way to break ammonia apart, sidestepping long‑standing limits in catalyst design and pointing toward cleaner, more efficient hydrogen production.
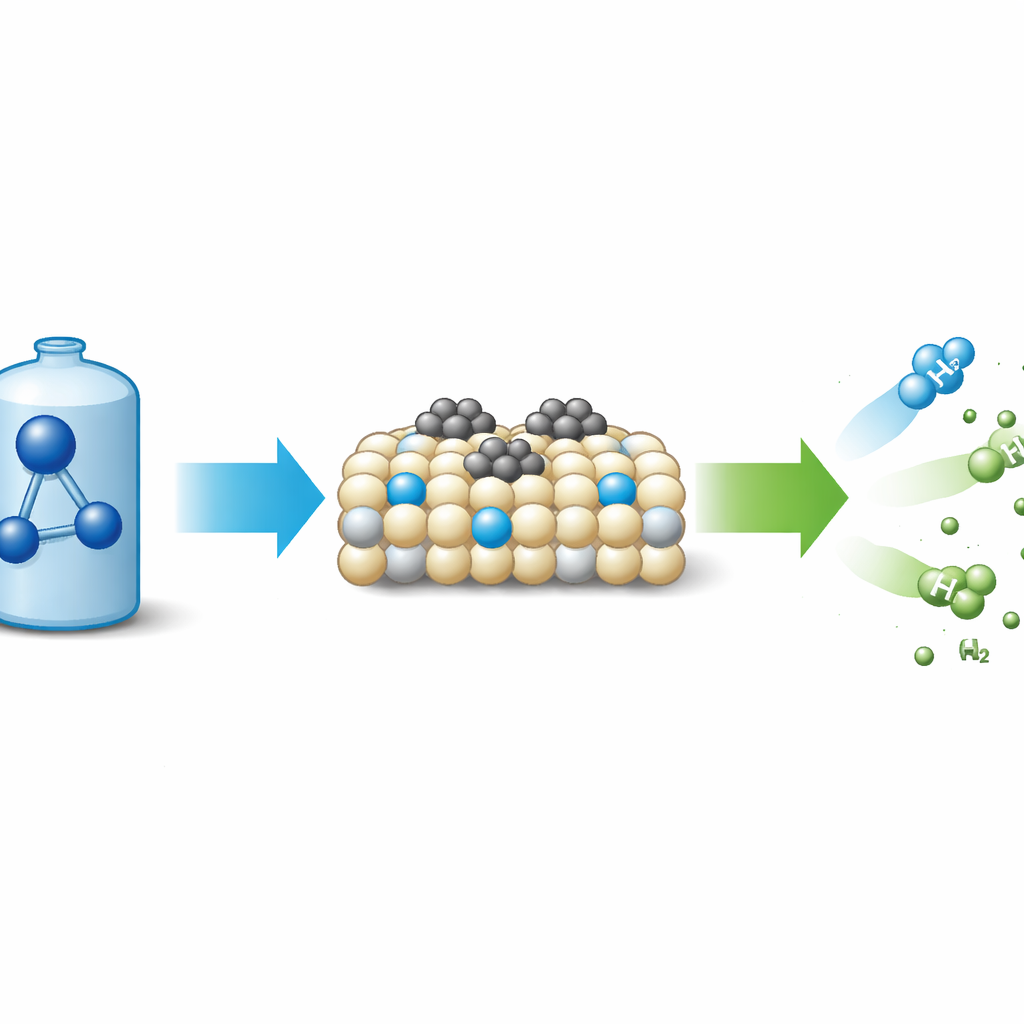
Why Ammonia Matters for Green Energy
Ammonia (NH₃) holds more hydrogen per liter than compressed hydrogen gas and is easy to liquefy and transport. When decomposed, it yields hydrogen and nitrogen—no carbon emissions. The challenge is that this reaction is energy‑hungry, so industry relies on powerful catalysts to lower the temperature and cost. Ruthenium, a precious metal, is currently the gold standard for speeding ammonia breakdown, but it follows a well‑known pattern: first the N–H bonds split on the metal surface, then nitrogen atoms pair up and leave as N₂. This “dissociative” route is trapped by a basic trade‑off: metals that grip nitrogen strongly help break N–H bonds but then hold on too tightly to nitrogen atoms, making it hard for N₂ to desorb. That trade‑off, captured in a relationship known as Brønsted–Evans–Polanyi scaling, has become a major bottleneck for better catalysts.
Borrowing a Trick from Nature
Nature’s nitrogen‑processing enzymes, called nitrogenases, avoid this trap by following a different script. Rather than tearing N≡N bonds apart in one go, they add and remove hydrogen stepwise at special redox‑active sites, forming and reshaping N–H and N–N bonds in a more cooperative way. Inspired by this, the authors asked whether a solid catalyst could mimic such “associative” behavior, where ammonia interacts with a reactive site that can both give and take electrons and temporarily store nitrogen. They focused on cerium oxide (CeO₂), a well‑known “oxygen shuttle” material whose atoms can easily switch between oxidation states. By inserting a small amount of nitrogen into the lattice—creating nitrogen‑doped cerium oxide—they aimed to turn some of the lattice sites into redox‑active nitrogen centers that might participate directly in breaking and forming nitrogen bonds.
Designing a Dual‑Site Catalyst
The team synthesized cerium oxide using a soft‑gel route and then treated it with flowing ammonia at various temperatures to introduce nitrogen atoms into the crystal. These nitrogen dopants generated extra defects and oxygen vacancies, making the surface more easily reduced and enhancing its electron‑donating power. When tiny ruthenium nanoparticles were deposited on this nitrogen‑doped support, the resulting catalyst (Ru/N‑CeO₂) decomposed ammonia far more efficiently than the same amount of ruthenium on undoped cerium oxide. At 450–500 °C, the optimized material reached nearly complete conversion with lower metal loading than many state‑of‑the‑art systems and remained stable for at least 70 hours. Measurements of reaction rates showed that the apparent activation energy dropped well below the typical range for ruthenium catalysts, a strong hint that the reaction pathway itself had changed.
Seeing a New Pathway in Action
To uncover what was really happening on the surface, the researchers combined infrared spectroscopy under operating conditions, isotopic labeling experiments, and advanced computer simulations. Infrared signals revealed the appearance of nitrogen–nitrogen and N–N–H–type species on the nitrogen‑doped support, suggesting that incoming ammonia molecules were forming bonds directly with lattice nitrogen atoms. Carefully timed pulses of ammonia containing heavier nitrogen (¹⁵N) showed that some of the nitrogen in the product N₂ came from these doped lattice sites, not just from the gas. Mass‑spectrometry data allowed the team to quantify contributions from two simultaneous routes: a conventional dissociative path on ruthenium and an associative, so‑called Mars–van Krevelen path in which lattice nitrogen temporarily leaves and is later refilled by nitrogen from ammonia. Roughly one‑third of the N₂ formed followed this new associative pathway.
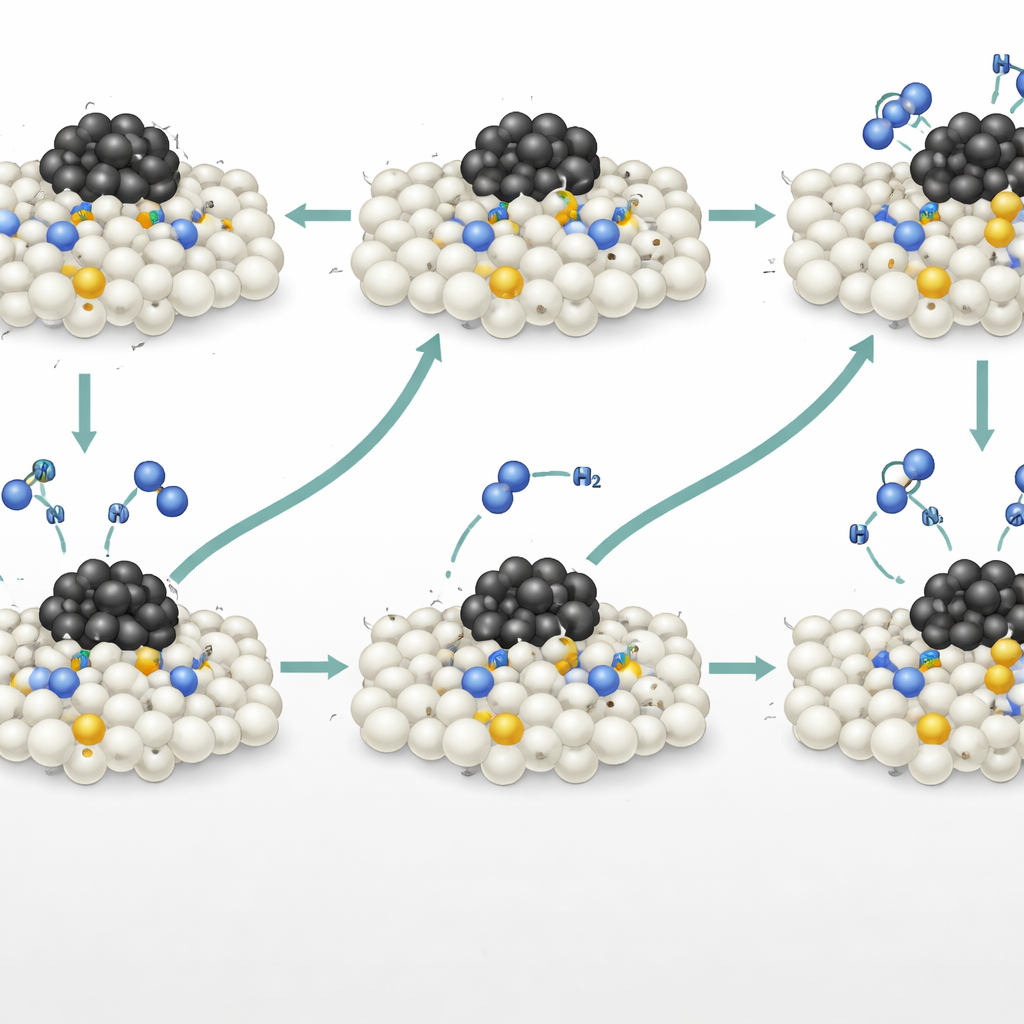
Where the Most Active Sites Live
Computer models based on machine‑learning‑accelerated simulations, checked against detailed quantum‑mechanical calculations, mapped the energetic landscape of this associative route. They showed that nitrogen dopants near—but not directly bonded to—the ruthenium particles offered the lowest overall energy barrier. These “proximal” sites feel the electronic influence of the metal strongly enough to ease N–H bond breaking, yet they are far enough away that nitrogen molecules can still depart without being stuck to ruthenium. In contrast, nitrogen right at the metal–support boundary binds too strongly and slows nitrogen release, while distant sites lack enough help from the metal. This non‑intuitive finding overturns the common belief that the metal–support interface is always the most active region, highlighting instead a sweet‑spot distance where lattice nitrogen acts as a powerful, independent active center.
A New Playbook for Hydrogen from Ammonia
By proving that nitrogen dopants in a redox‑active support can act as their own catalytic sites, working alongside metal nanoparticles but through a distinct mechanism, this work shows a concrete route to break free of long‑standing scaling limits in ammonia decomposition. Carefully positioning and stabilizing these lattice nitrogen sites allows ammonia to be broken down through an associative pathway that couples N–H bond scission with N–N bond formation, lowering energy demands and enabling high activity at milder conditions. For non‑specialists, the takeaway is that the “background” support in a catalyst is not just an inert scaffold: when chemically engineered, it can share the workload with precious metals and even reshape the reaction itself. That insight could guide the design of next‑generation catalysts for ammonia‑based hydrogen systems and other clean‑energy reactions that currently seem hemmed in by fundamental trade‑offs.
Citation: Ye, D., Luo, M., Liu, X. et al. Unravelling the role of redox active sites in nitrogen doped cerium oxide for associative ammonia decomposition. Nat Commun 17, 3892 (2026). https://doi.org/10.1038/s41467-026-70330-5
Keywords: ammonia decomposition, hydrogen production, ruthenium catalyst, nitrogen-doped ceria, heterogeneous catalysis