Clear Sky Science · sv
Avkodning av lokal ramverksdynamik i ultra-små porer i MOF:en MIL-120(Al) CO2-adsorbent med maskininlärningspotential
Varför små porer och rörliga atomer spelar roll för koldioxidavskiljning
Att stoppa klimatförändringen kommer sannolikt att kräva att koldioxid (CO2) fångas in från industriella utsläpp och möjligen även från luften. En lovande materialklass för detta är metall–organiska ramverk — kristalllika stommar fulla av nanoskopiska porer som kan fånga gasmolekyler. I den här studien undersöks ett särskilt ramverk kallat MIL-120(Al), som har extremt smala kanaler. Forskarna visar att mycket små, snabba rörelser hos vätebärande grupper inne i dessa porer, osynliga för de flesta experiment, starkt kan påverka hur väl materialet fångar CO2. Med kvantberäkningar och en skräddarsydd maskininlärningsmodell kartlägger de hur dessa dolda rörelser styr både var CO2 placerar sig i porerna och hur starkt det binds.
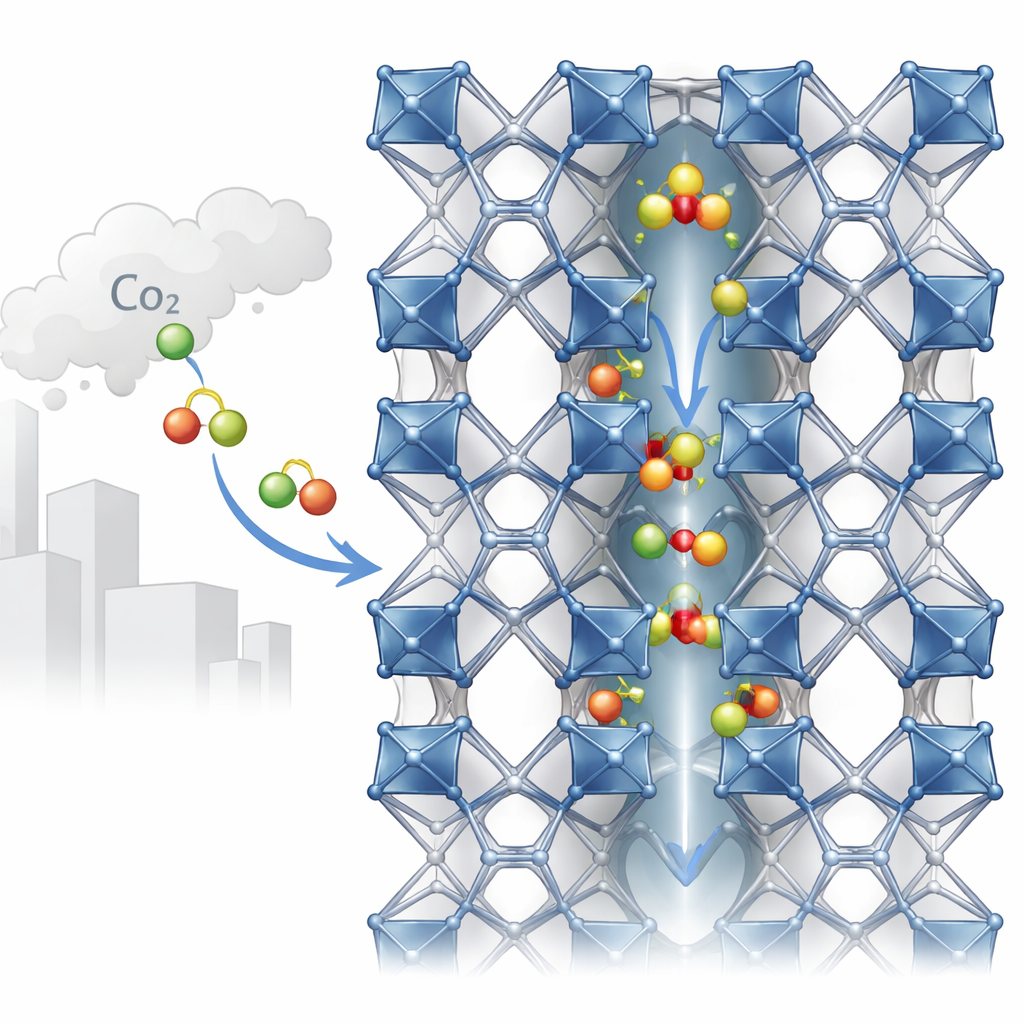
Små tunnlar byggda av metaller och organiska delar
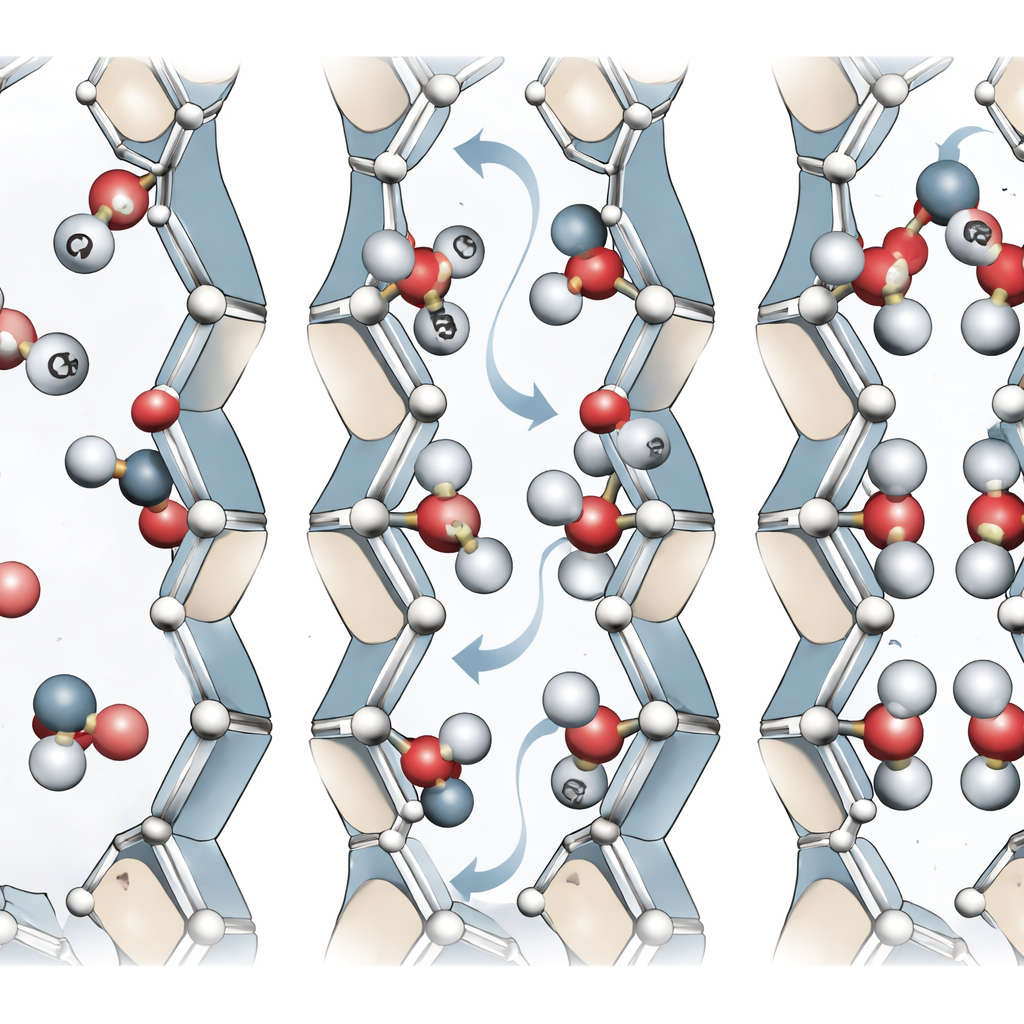
MIL-120(Al) byggs upp av aluminiumatomer kopplade med en organisk molekyl till ett robust tredimensionellt ramverk. Denna arkitektur skapar endimensionella kanaler som är endast omkring en halv nanometer breda — knappt tillräckligt utrymme för gasmolekyler som CO2 att passera. Längs väggarna i dessa kanaler sitter så kallade “bryggande hydroxylgrupper”: små enheter som inkluderar en väteatom bunden till syre, som förbinder aluminiumcentra. Experiment med röntgen svårigheter att se väteatomer, så tidigare strukturella modeller gissade ofta positionerna för dessa grupper. Det nya arbetet utmanar den antagandet genom att visa att hur dessa små grupper pekar — mot en närliggande kedja eller in i kanalen — subtilt ändrar den effektiva porebredden och sättet som CO2 binds.
Många dolda arrangemang i ett enda material
Genom kvantmekaniska (DFT) beräkningar kartlade teamet sex distinkta sätt som dessa hydroxylgrupper kan vara arrangerade i enhetscellen för MIL-120(Al). Även om alla sex versioner ser nästan identiska ut i röntgenexperiment och har mycket lika cellmått, skiljer de sig i energi och poreform. Den lägstenergetiska versionen bildar ett intrikat nätverk av vätebindningar mellan närliggande kedjor, medan andra har mer oordnade eller mer porevända hydroxylgrupper. Dessa skillnader ändrar kanalstorleken med mindre än en ångström, men i så här ultrasmala porer kan även sub-ångströmförändringar drastiskt påverka vilka gasmolekyler som kan tränga in och hur hårt de är inneslutna. Författarna identifierar därför hydroxylorientering som en “dold” strukturell variabel som standardkarakterisering förbiser.
Maskininlärning för att följa rörliga atomer
För att följa hur dessa hydroxylgrupper rör sig och växlar mellan arrangemang tränade forskarna en dedikerad maskininlärningspotential på en stor mängd högkvalitativa kvantdata. Denna modell reproducerar energier, krafter, vibrationer och övergångsvägar med nära kvantnivåns noggrannhet men till en bråkdel av beräkningskostnaden. Med den kunde de utforska hur lätt de olika arrangemangen kan omväxlas. Energibarriärerna är låga, vilket betyder att vid rumstemperatur kan hydroxylgrupperna flippa mellan konfigurationer snarare än vara frusna. Mekaniska tester baserade på samma modell visar att även om materialets övergripande styvhet är likartad mellan arrangemangen, beror dess respons på belastning i olika riktningar starkt på hur dessa grupper är ordnade. Detta avslöjar ett flexibelt men robust ramverk vars interna kontakter styrs av vätebindningar.
Hur CO2 hittar sin plats i porerna
Med sin maskininlärningsmodell simulerade författarna hur CO2-molekyler går in och ordnar sig inne i MIL-120(Al). De upptäckte att CO2 tenderar att antingen ställa in sig tvärs över kanalen eller längs den, beroende på hur hydroxylgrupperna pekar. När hydroxylerna vänder in i poren kan CO2 bilda starka vätebindningsliknande kontakter, vilket leder till högre bindningsenergier och något högre adsorptionvärme. När de är mer axiallyt ordnade, radar CO2 i stället upp längs kanalen. Avancerade simuleringar som tillåter både gasen och ramverket att röra sig — snarare än att behandla ramverket som styvt — återger experimentell CO2-upptagning och bindningsstyrkor mycket bättre än standard kraftfältsmetoder. Resultaten visar också att när fler CO2 fyller porerna kan detta påskynda omorienteringen av hydroxylgrupper, vilket hjälper ramverket att omorganisera sig till energimässigt fördelaktiga tillstånd.
Vad detta betyder för framtida CO2-filter
Sammanfattningsvis visar studien att småskaliga rörelser av väteatomer i MIL-120(Al) inte är en marginell detalj utan en central faktor som styr hur CO2 fångas. Även om röntgenmätningar inte tydligt kan se dessa väten, justerar deras skiftande positioner poreöppningen samt styrka och geometri för CO2-bindning. Genom att kombinera precisa kvantberäkningar med en systemspecifik maskininlärningspotential bygger författarna en realistisk bild av denna dolda interna dynamik och dess påverkan på gasadsorption. För konstruktörer av nästa generations CO2-filter och relaterade material är budskapet tydligt: för att förutsäga och optimera prestanda i ultrasmå porer är det avgörande att ta hänsyn till den subtila dansen hos interna funktionella grupper, inte bara det genomsnittliga, stela ramverket.
Citering: Fan, D., Oliveira, F.L., Bonakala, S. et al. Decoding local framework dynamics in the ultra-small pore MOF MIL-120(Al) CO2 adsorbent using machine-learning potential. Nat Commun 17, 3235 (2026). https://doi.org/10.1038/s41467-026-69993-x
Nyckelord: metall-organiska ramverk, koldioxidfångst, ultra-mikroporösa material, maskininlärningspotentialer, gasadsorption