Clear Sky Science · fr
Déchiffrer la dynamique locale du réseau dans l’adsorbant CO2 MOF à pores ultra-petits MIL-120(Al) à l’aide d’un potentiel d’apprentissage automatique
Pourquoi les pores minuscules et les atomes en mouvement comptent pour la capture du carbone
Arrêter le changement climatique exigera probablement d’extraire le dioxyde de carbone (CO2) des fumées industrielles et peut‑être même de l’air ambiant. Une classe de matériaux prometteuse pour cette tâche est celle des cadres métal–organiques : des architectures cristallines ponctuées de pores nanoscalaires capables de piéger des molécules de gaz. Cette étude porte sur un réseau particulier, MIL-120(Al), qui présente des canaux extrêmement étroits. Les chercheurs montrent que de très petits mouvements rapides de groupes porteurs d’hydrogène à l’intérieur de ces pores, invisibles à la plupart des expériences, peuvent influencer fortement l’efficacité de capture du CO2. En combinant calculs quantiques et un modèle d’apprentissage automatique adapté, ils dévoilent comment ces mouvements cachés contrôlent à la fois les positions privilégiées du CO2 dans les pores et la force d’adsorption.
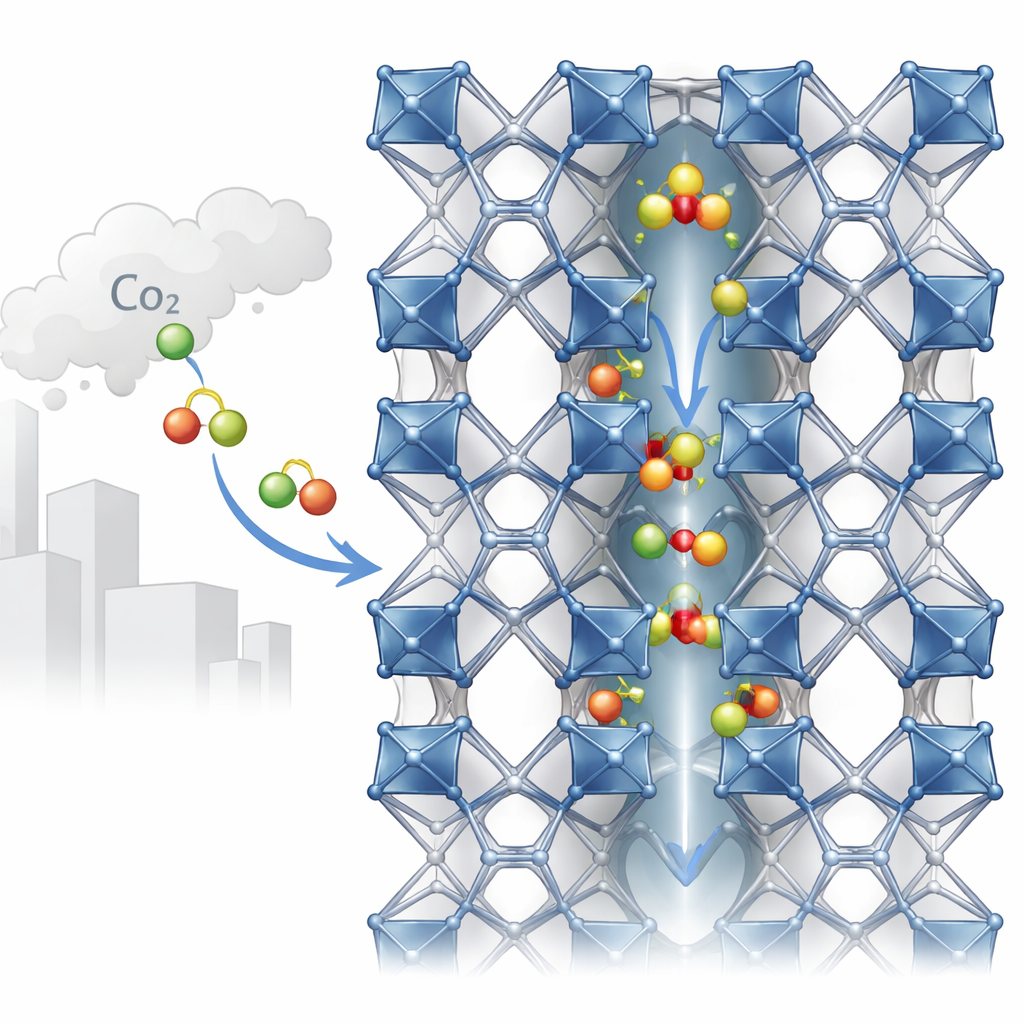
Des tunnels minuscules construits à partir de métaux et d’éléments organiques
MIL-120(Al) est construit à partir d’atomes d’aluminium reliés par une molécule organique en un réseau tridimensionnel robuste. Cette architecture crée des canaux unidimensionnels d’environ un demi‑nanomètre de largeur — à peine assez d’espace pour que des molécules de gaz comme le CO2 les traversent. Le long des parois de ces canaux se trouvent des « hydroxyles pontants » : de petites unités comprenant un atome d’hydrogène lié à l’oxygène, connectant les centres d’aluminium. Les expériences par rayons X ont du mal à localiser les hydrogènes, si bien que les modèles structuraux antérieurs ont simplement supposé leurs positions. Le travail présent remet en question cette hypothèse en montrant que l’orientation de ces petits groupes — vers une chaîne voisine ou vers le canal — modifie subtilement la largeur effective du pore et la manière dont le CO2 y est accueilli.
De nombreuses configurations cachées au sein d’un même matériau
À l’aide de calculs de mécanique quantique (DFT), l’équipe a cartographié six configurations distinctes possibles pour ces groupes hydroxyles dans la maille répétitive de MIL-120(Al). Bien que les six versions apparaissent presque identiques aux expérimentations par rayons X et présentent des dimensions cellulaires globales très proches, elles diffèrent par leur énergie et la forme des pores. La configuration la plus stable en énergie forme un réseau imbriqué de liaisons hydrogène entre chaînes voisines, tandis que d’autres présentent des hydroxyles plus désordonnés ou orientés vers le pore. Ces différences modifient la taille du canal de moins d’un angström, mais dans de tels pores ultra‑petits, des variations sub‑ångström peuvent énormément influencer quelles molécules de gaz peuvent pénétrer et à quel point elles sont confinées. Les auteurs identifient donc l’orientation des hydroxyles comme une variable structurelle « cachée » que la caractérisation standard néglige.
Apprentissage automatique pour suivre les atomes en mouvement
Pour suivre la dynamique de ces groupes hydroxyles et leurs basculements entre configurations, les chercheurs ont entraîné un potentiel d’apprentissage automatique dédié sur un large jeu de données quantiques de haute précision. Ce modèle reproduit les énergies, forces, vibrations et voies de transition avec une précision proche de la méthode quantique, mais à une fraction du coût informatique. Grâce à lui, ils ont pu explorer la facilité d’interconversion entre configurations. Les barrières énergétiques sont faibles, ce qui signifie qu’à température ambiante les hydroxyles peuvent basculer entre états plutôt que d’être figés. Des tests mécaniques basés sur le même modèle montrent que, bien que la rigidité globale du matériau soit similaire selon les configurations, la réponse au stress selon différentes directions dépend fortement de l’ordre de ces groupes. Cela révèle un réseau flexible mais robuste dont les contacts internes sont réglés par les liaisons hydrogène.
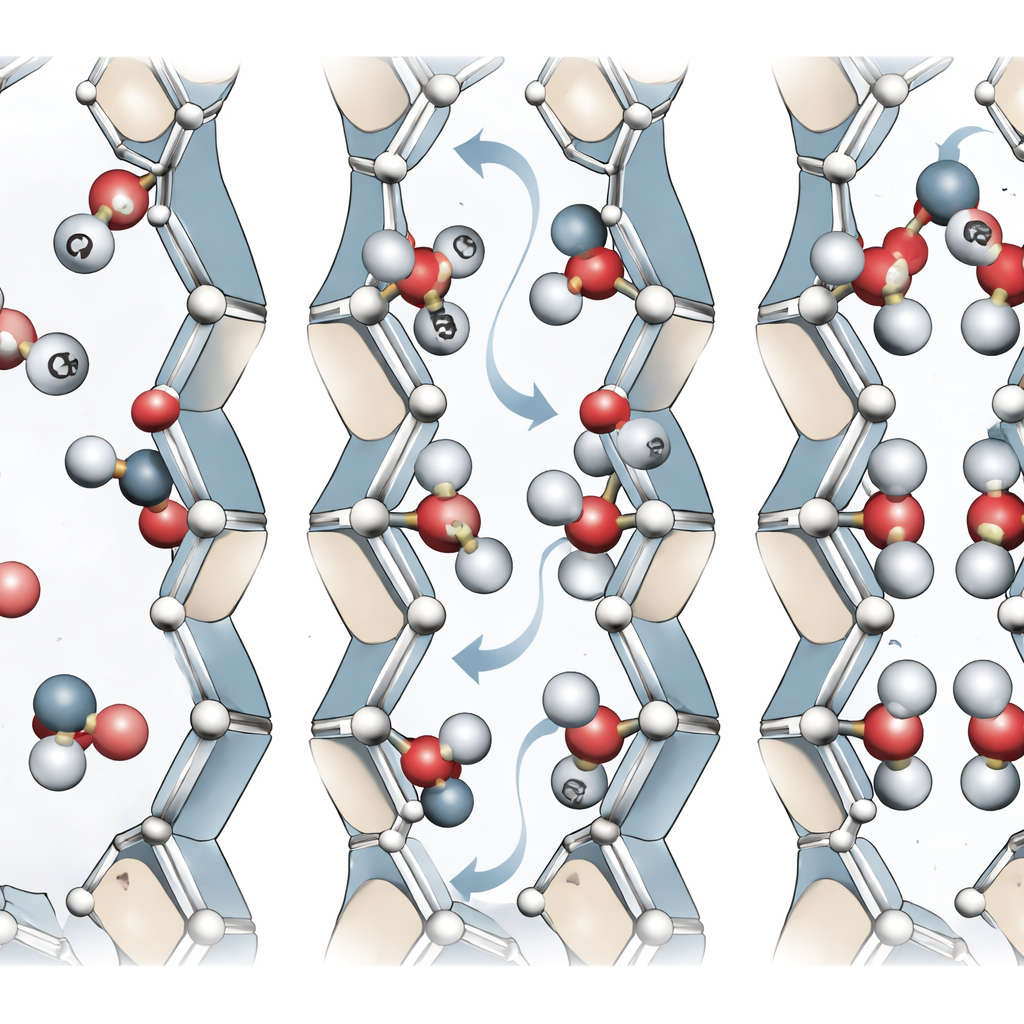
Comment le CO2 trouve sa place dans les pores
Avec leur modèle d’apprentissage automatique, les auteurs ont simulé la manière dont les molécules de CO2 pénètrent et se disposent à l’intérieur de MIL-120(Al). Ils ont observé que le CO2 tend à s’aligner soit transversalement au canal, soit selon l’axe du canal, selon l’orientation des hydroxyles. Quand les hydroxyles font face à l’intérieur du pore, le CO2 peut établir des contacts de type liaison hydrogène plus forts, conduisant à des énergies de liaison plus élevées et à une chaleur d’adsorption légèrement supérieure. Lorsqu’ils sont orientés plus axialement, le CO2 s’aligne le long du canal. Des simulations avancées permettant au gaz et au réseau de se mouvoir — plutôt que de traiter le réseau comme rigide — reproduisent l’adsorption et les forces de liaison expérimentales du CO2 bien mieux que les approches classiques par champs de force. Les résultats montrent aussi qu’à mesure que les pores se remplissent de CO2, cela peut accélérer la réorientation des hydroxyles, aidant le réseau à se réorganiser en états énergétiquement favorables.
Ce que cela signifie pour les filtres à CO2 de demain
Dans l’ensemble, l’étude révèle que les mouvements à petite échelle des atomes d’hydrogène dans MIL-120(Al) ne sont pas un détail mineur mais un facteur central contrôlant la capture du CO2. Même si les mesures par rayons X ne distinguent pas clairement ces hydrogènes, leurs positions changeantes ajustent l’ouverture des pores ainsi que la force et la géométrie de liaison du CO2. En combinant des calculs quantiques précis avec un potentiel d’apprentissage automatique spécifique au système, les auteurs construisent une image réaliste de cette dynamique interne cachée et de son impact sur l’adsorption des gaz. Pour les concepteurs de filtres CO2 de nouvelle génération et matériaux apparentés, le message est clair : pour prédire et optimiser les performances dans des pores ultra‑petits, il est crucial de prendre en compte la danse subtile des groupes fonctionnels internes, et pas seulement le réseau moyen et rigide.
Citation: Fan, D., Oliveira, F.L., Bonakala, S. et al. Decoding local framework dynamics in the ultra-small pore MOF MIL-120(Al) CO2 adsorbent using machine-learning potential. Nat Commun 17, 3235 (2026). https://doi.org/10.1038/s41467-026-69993-x
Mots-clés: matériaux métal–organique (MOF), captation du dioxyde de carbone, matériaux ultra-microporeux, potentiels par apprentissage automatique, adsorption des gaz