Clear Sky Science · en
Decoding local framework dynamics in the ultra-small pore MOF MIL-120(Al) CO2 adsorbent using machine-learning potential
Why tiny pores and moving atoms matter for carbon capture
Stopping climate change will likely require pulling carbon dioxide (CO2) out of industrial exhaust and possibly even out of the air. One promising class of materials for this job is metal–organic frameworks—crystal-like scaffolds full of nanoscale pores that can trap gas molecules. This study looks at a particular framework called MIL-120(Al), which has extremely narrow channels. The researchers show that very small, fast motions of hydrogen-bearing groups inside these pores, invisible to most experiments, can strongly influence how well the material captures CO2. Using quantum calculations and a tailored machine-learning model, they uncover how these hidden motions control both where CO2 sits in the pores and how strongly it sticks.
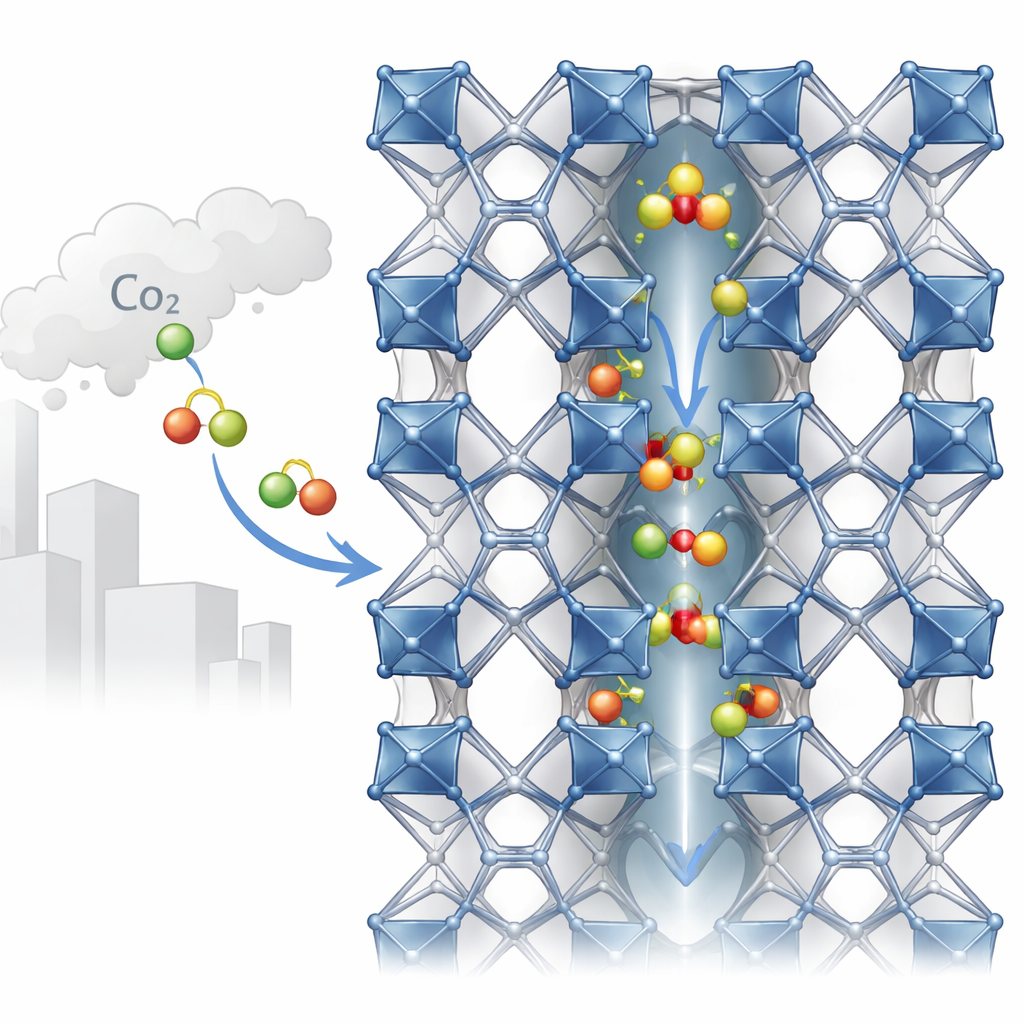
Tiny tunnels built from metals and organic pieces
MIL-120(Al) is built from aluminum atoms linked by an organic molecule into a sturdy three-dimensional framework. This architecture creates one-dimensional channels only about half a nanometer wide—barely enough space for gas molecules like CO2 to pass through. Along the walls of these channels sit “bridging hydroxyl” groups: small units that include a hydrogen atom attached to oxygen, connecting aluminum centers. Experiments using X-rays have difficulty seeing hydrogen atoms, so past structural models simply guessed the positions of these groups. The new work challenges that assumption by showing that how these tiny groups point—toward a neighboring chain or into the channel—subtly changes the effective pore width and the way CO2 is hosted.
Many hidden arrangements inside one material
Using quantum mechanical (DFT) calculations, the team mapped out six distinct ways these hydroxyl groups can be arranged in the repeating unit of MIL-120(Al). Although all six versions look nearly identical to X-ray experiments and have very similar overall cell dimensions, they differ in energy and pore shape. The lowest-energy version forms an interlocking network of hydrogen bonds between neighboring chains, while others have more disordered or more pore-facing hydroxyl groups. These differences shift the channel size by less than an ångström, but in such ultra-small pores even sub-angstrom changes can drastically affect which gas molecules can enter and how tightly they are confined. The authors therefore identify hydroxyl orientation as a “hidden” structural variable that standard characterization overlooks.
Machine learning to follow moving atoms
To track how these hydroxyl groups move and switch between arrangements, the researchers trained a dedicated machine-learning potential on a large set of high-level quantum data. This model reproduces energies, forces, vibrations, and transition pathways with near-quantum accuracy but at a tiny fraction of the computational cost. With it, they could explore how easily the different arrangements interconvert. The energy barriers are low, meaning that at room temperature the hydroxyl groups can flip between configurations rather than being frozen. Mechanical tests based on the same model show that, although the overall stiffness of the material is similar across arrangements, the way it responds to stress in different directions depends strongly on how these groups are ordered. This reveals a flexible yet robust framework whose internal contacts are tuned by hydrogen bonding.
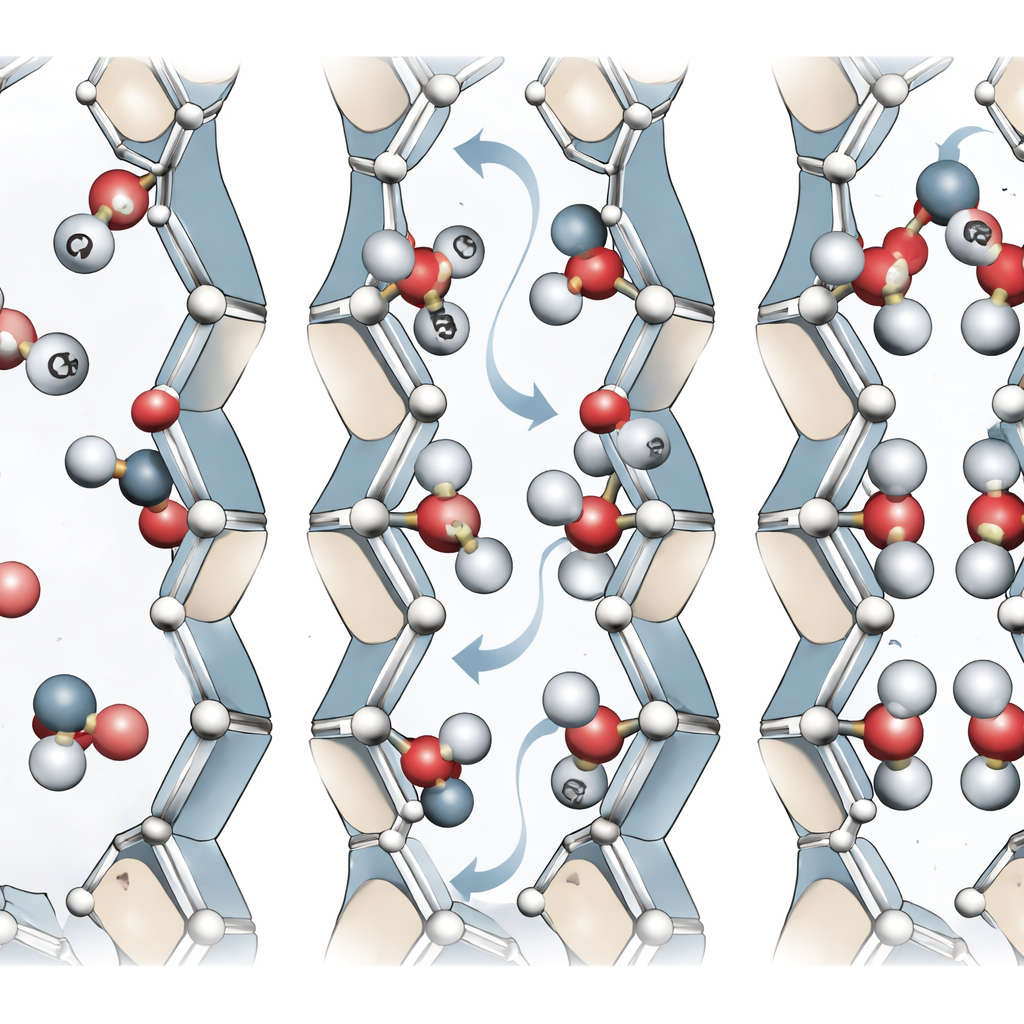
How CO2 finds its place in the pores
With their machine-learning model, the authors simulated how CO2 molecules enter and arrange themselves inside MIL-120(Al). They discovered that CO2 tends to align either roughly across the channel or along it, depending on how the hydroxyl groups point. When hydroxyls face into the pore, CO2 can form strong hydrogen-bond-like contacts, leading to higher binding energies and slightly higher heat of adsorption. When they are arranged more axially, CO2 lines up along the channel instead. Advanced simulations that allow both the gas and the framework to move—rather than treating the framework as rigid—reproduce experimental CO2 uptake and binding strengths much better than standard force-field approaches. The results also show that as more CO2 fills the pores, it can accelerate the reorientation of hydroxyl groups, helping the framework reorganize into energetically favorable states.
What this means for future CO2 filters
Overall, the study reveals that small-scale motions of hydrogen atoms in MIL-120(Al) are not a minor detail but a central factor controlling how CO2 is captured. Even though X-ray measurements cannot see these hydrogens clearly, their shifting positions adjust the pore opening and the strength and geometry of CO2 binding. By combining accurate quantum calculations with a system-specific machine-learning potential, the authors build a realistic picture of this hidden internal dynamics and its impact on gas adsorption. For designers of next-generation CO2 filters and related materials, the message is clear: to predict and optimize performance in ultra-small pores, it is crucial to account for the subtle dance of internal functional groups, not just the average, rigid framework.
Citation: Fan, D., Oliveira, F.L., Bonakala, S. et al. Decoding local framework dynamics in the ultra-small pore MOF MIL-120(Al) CO2 adsorbent using machine-learning potential. Nat Commun 17, 3235 (2026). https://doi.org/10.1038/s41467-026-69993-x
Keywords: metal-organic frameworks, carbon dioxide capture, ultra-microporous materials, machine-learning potentials, gas adsorption