Clear Sky Science · sv
Från enkelsekvenser till evolutionära banor: protein‑språkmodeller fångar SARS‑CoV‑2:s evolutionära potential
Varför detta är viktigt för framtida pandemier
Under större delen av COVID‑19‑pandemin låg forskarna efter: nya varianter dök upp i verkligheten innan laboratorier hann avgöra vad förändringarna betydde för smittsamhet eller immunflykt. Den här studien visar att kraftfulla datormodeller, ursprungligen framtagna för att förstå mänskligt språk, i stället kan läsa proteiners ”språk” och härleda hur koronavirusets spike‑protein sannolikt kommer att förändras och anpassa sig — endast utifrån dess byggstenars sekvens. Den förmågan kan hjälpa forskare att tidigare flagga oroande varianter och kan generaliseras till många andra patogener.
Att lära datorer att läsa proteiner
Författarna arbetar med en protein‑språkmodell kallad ESM‑2, tränad på tiotals miljoner proteinsekvenser från hela livets träd. Precis som en språkmodell lär sig grammatik och betydelse ur ord lär sig ESM‑2 vilka aminosyra‑mönster som ”är rimliga” i verkliga proteiner. När modellen får sekvensen för SARS‑CoV‑2‑spiken tilldelar den varje möjlig mutation två nyckelskalor: en grammatikalitetspoäng som återspeglar hur väl en förändrad sekvens passar de inlärda reglerna för proteinstruktur, och en semantisk poäng som mäter hur annorlunda hela proteinet blir i modellens interna representation. Dessa poäng kan beräknas för varje möjlig enstaka mutation i silico, en strategi som kallas in silico djup mutageneskanning. 
Kartlägga var viruset kan och inte kan förändras
Genom att skanna alla enstaka bokstavsändringar över spike fann teamet att ESM‑2 naturligt återfinner proteinets viktigaste arkitektoniska drag. S2‑delen, som bildar den stabila stammen som driver membranfusion, förutsägs vara starkt begränsad: de flesta mutationer där sänker grammatikaliteten markant, vilket antyder att de skulle skada proteinets struktur eller funktion. I kontrast tolererar yttre regioner av S1, inklusive N‑terminala domänen och receptorbindande domänen, mycket fler förändringar. Detta stämmer med vad som ses i verkliga virusgenom, där dessa exponerade regioner ackumulerar mutationer som hjälper viruset fästa vid celler och undvika antikroppar medan den strukturella kärnan förblir mer konservativ.
Upptäcka dold samverkan mellan mutationer
Proteiner är inte bara en uppsättning oberoende platser; en mutation kan ändra hur andra mutationer accepteras, ett fenomen kallat epistas. Forskarna undersökte detta genom att utgå från Omicron BA.1‑spiken och i datorn ”återställa” dess karakteristiska mutationer en i taget tillbaka till den ursprungliga Wuhan‑sekvensen. Varje återställning ändrar modellens sannolikheter för aminosyror vid alla andra positioner. Stora skift avslöjar par av platser vars öden är kopplade. Med denna metod lyfter studien fram kända hotspots såsom positioner kring 484 och 501 i receptorbindande domänen, som tillsammans påverkar både immunflykt och bindning till ACE2‑receptorn. Den pekar också på mindre uppenbara kluster av rester vars interaktioner senare bekräftades i experimentella studier av Omicrons förbättrade tillväxt i mänskliga näsceller, vilket tyder på att modellen fångar äkta strukturella och funktionella kopplingar. 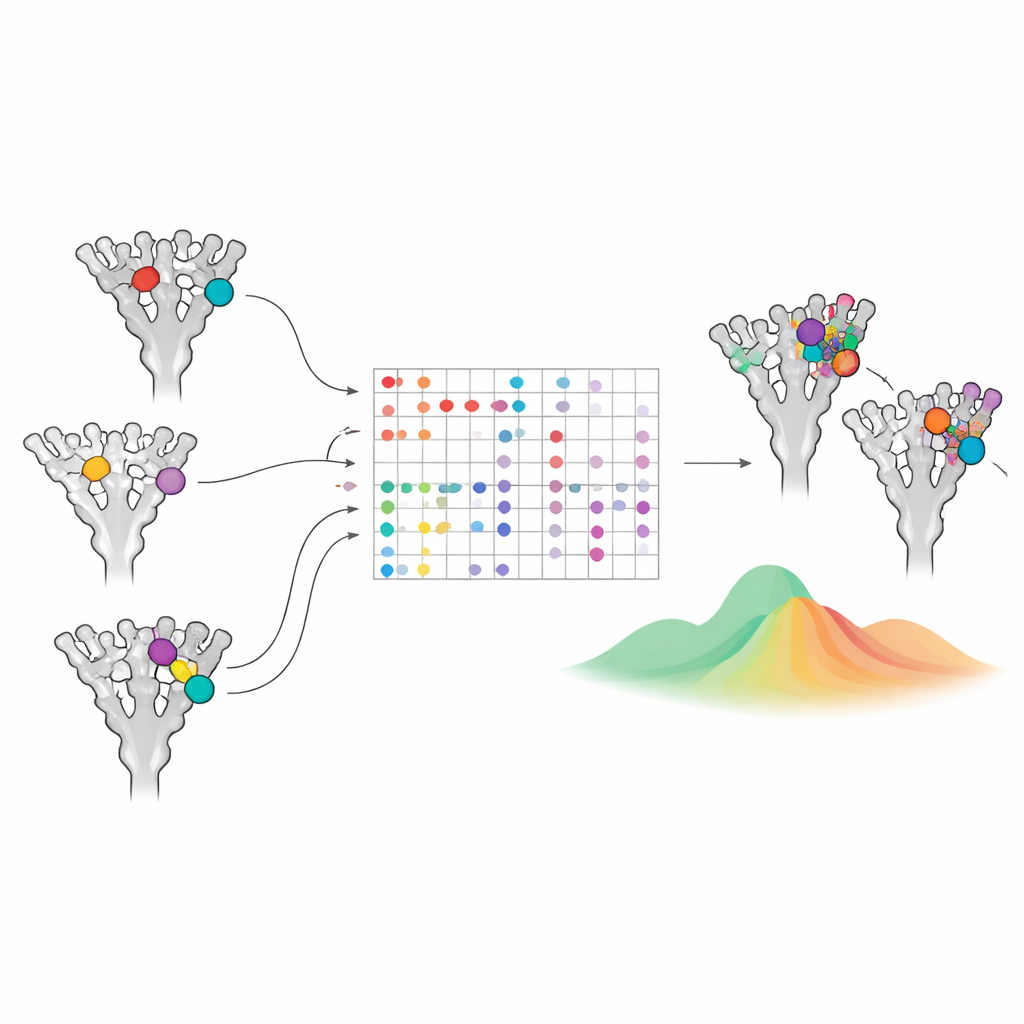
Följa virusets evolution och upptäcka avvikare
Bortom enstaka mutationer frågar författarna om ESM‑2 kan tolka hela variantsekvenser när de dyker upp över tid. De bäddar in en spiksekvens för varje namngiven SARS‑CoV‑2‑linje och placerar dem i en tvådimensionell karta med en metod som kallas evo‑velocity, vilken även härleder en dominerande riktning för förändring. Den resulterande kartläggningen speglar det kända fylogenetiska trädet: tidiga linjer klustrar ihop sig, och sedan skär sig grenar som motsvarar Alpha, Delta, Omicron och rekombinanta linjer av i rätt tidsordning. Enkla sammanfattande statistiker såsom genomsnittlig grammatikalitet och semantiskt avstånd separerar tydligt icke‑variantlinjer, tidiga intressanta varianter och Omicron‑klassvirus, vilket visar att modellens interna representation spårar meningsfulla evolutionära skiften.
Göra inbäddningar till ett tidigt varningssystem
För att utforska praktisk övervakning introducerar teamet en dynamisk semantisk poäng: varje ny spiksekvens jämförs inte bara med ursprungsstammen från Wuhan utan med genomsnittet av virus som cirkulerade under de föregående tre månaderna. Tillämpad på täta sekvenseringsdata från Storbritannien ger denna rörliga poäng distinkta vågor i takt med att Alpha, Delta och efterföljande Omicron‑sublinjer stiger och faller. Sekvenser som ligger en eller två standardavvikelser från det aktuella genomsnittet flaggas som potentiella sekvenser av oro. Med enbart dessa tidiga avvikare skulle metoden ha lyft fram de flesta av Världshälsoorganisationens varianter av oro och flera viktiga senare avknoppningar såsom JN.1, samtidigt som den visar vilka specifika platser i spike‑proteinet som upprepade gånger ändras i framväxande linjer.
Vad detta innebär för framtida hot
Sammanfattningsvis visar studien att en allmän protein‑språkmodell, använd direkt utan specialanpassning, kan identifiera vilka delar av SARS‑CoV‑2‑spiken som är flexibla, vilka platser som är strukturellt kritiska, hur mutationer samspelar och hur virusets spike vandrat genom evolutionärt rum under pandemins gång. Eftersom metoden fungerar från en enda proteinsekvens och inte är beroende av föreliggande alignments eller detaljerade strukturdatasätt, kan den tillämpas mycket tidigt i ett utbrott, när bara ett fåtal genom är kända. I takt med att liknande modeller förfinas och anpassas till virala dataset kan de bli en viktig del av verktygslådan för att förutsäga hur nya patogener utvecklas och för att prioritera varianter för laboratoriestudier och vaccinutveckling.
Citering: Lamb, K.D., Hughes, J., Lytras, S. et al. From single-sequences to evolutionary trajectories: protein language models capture the evolutionary potential of SARS-CoV-2. Nat Commun 17, 2938 (2026). https://doi.org/10.1038/s41467-026-69569-9
Nyckelord: protein‑språkmodeller, SARS‑CoV‑2 spike, virusets evolution, epistas, variantövervakning