Clear Sky Science · pt
De sequências únicas a trajetórias evolutivas: modelos de linguagem de proteínas capturam o potencial evolutivo do SARS-CoV-2
Por que isso importa para pandemias futuras
Na maior parte da pandemia de COVID‑19, os cientistas ficaram um passo atrás: novas variantes surgiam no mundo real antes que os laboratórios pudessem medir o que aquelas mudanças significavam para infectividade ou escape imune. Este estudo mostra que poderosos modelos computacionais, originalmente projetados para entender a linguagem humana, podem em vez disso ler a “linguagem” das proteínas e inferir como a proteína spike do coronavírus tende a mudar e se adaptar — usando apenas sua sequência de blocos de construção. Essa capacidade poderia ajudar pesquisadores a sinalizar variantes preocupantes mais cedo e pode se generalizar para muitos outros patógenos.
Ensinando computadores a ler proteínas
Os autores trabalham com um modelo de linguagem de proteínas chamado ESM‑2, treinado com dezenas de milhões de sequências de proteínas de toda a árvore da vida. Assim como um modelo de linguagem aprende gramática e significado a partir de palavras, o ESM‑2 aprende quais padrões de aminoácidos “fazem sentido” em proteínas reais. Ao receber a sequência da proteína spike do SARS‑CoV‑2, o modelo atribui a cada mutação possível duas pontuações-chave: uma pontuação de gramaticalidade que reflete quão bem uma sequência alterada se encaixa nas regras aprendidas da estrutura proteica, e uma pontuação semântica que mede o quão diferente a proteína se torna na representação interna do modelo. Essas pontuações podem ser calculadas para todas as mutações únicas possíveis em um computador, uma estratégia conhecida como varredura profunda de mutações in silico. 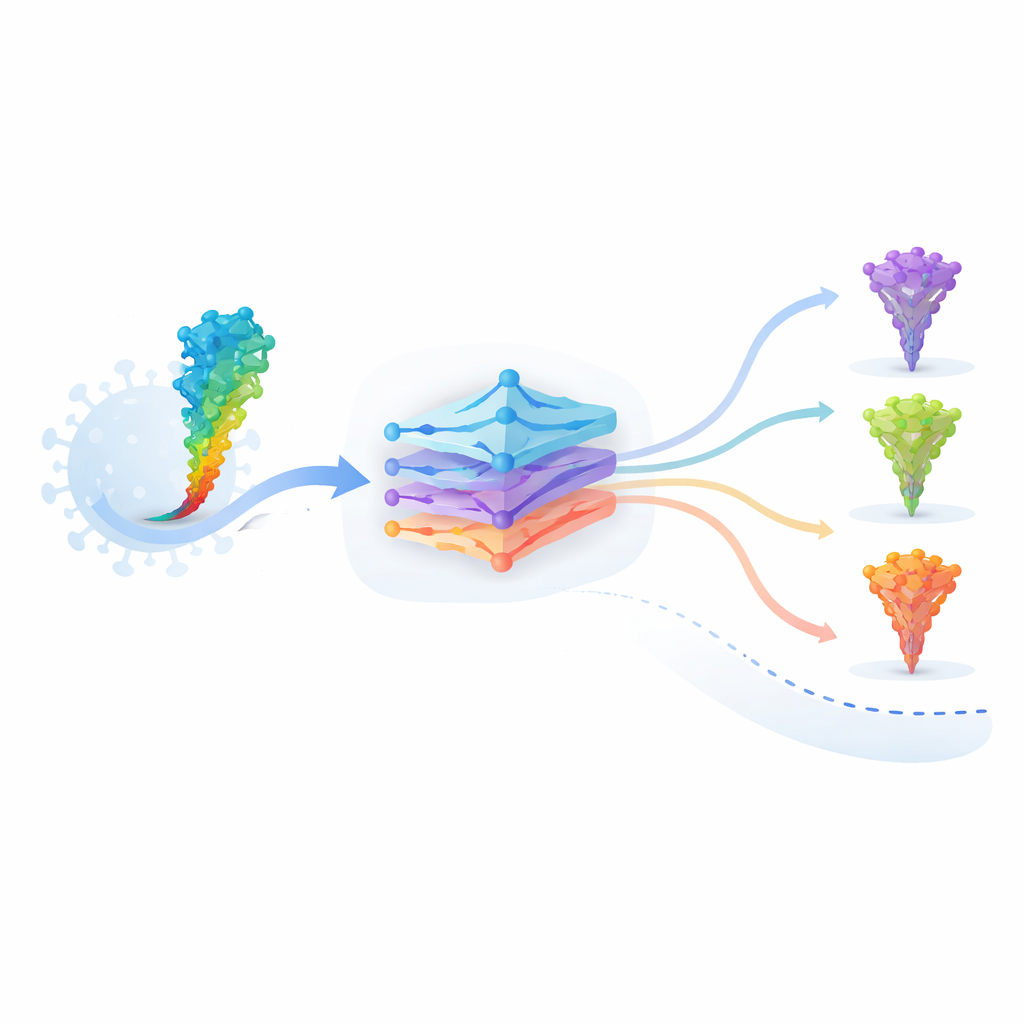
Mapeando onde o vírus pode e não pode mudar
Ao examinar todas as mudanças de uma letra ao longo da spike, a equipe descobriu que o ESM‑2 recupera naturalmente as principais características arquitetônicas da proteína. A porção S2, que forma o eixo estável que promove a fusão de membranas, é prevista como altamente conservada: a maioria das mutações ali reduz fortemente a gramaticalidade, implicando que danificariam a estrutura ou função da proteína. Em contraste, regiões na superfície externa da porção S1, incluindo o domínio N-terminal e o domínio de ligação ao receptor, toleram muito mais mudanças. Isso corresponde ao que se observa em genomas virais reais, onde essas regiões expostas acumulam mutações que ajudam o vírus a se ligar às células e escapar de anticorpos, enquanto o núcleo estrutural permanece mais conservado.
Revelando trabalho em equipe oculto entre mutações
Proteínas não são apenas um conjunto de sítios independentes; uma mutação pode alterar quão aceitáveis outras são, fenômeno chamado epistasia. Os pesquisadores investigaram isso começando pela spike Omicron BA.1 e “revertendo” computacionalmente suas mutações definidoras uma a uma de volta à sequência original de Wuhan. Cada reversão altera as probabilidades do modelo para aminoácidos em todas as outras posições. Grandes mudanças revelam pares de sítios cujos destinos estão ligados. Usando essa abordagem, o estudo destaca pontos conhecidos como posições ao redor de 484 e 501 no domínio de ligação ao receptor, que em conjunto moldam tanto o escape imune quanto a ligação ao receptor ACE2. Aponta também para agrupamentos menos óbvios de resíduos cujas interações foram posteriormente confirmadas em estudos experimentais do crescimento aumentado do Omicron em células nasais humanas, sugerindo que o modelo está capturando acoplamentos estruturais e funcionais genuínos. 
Acompanhando a evolução viral e identificando outliers
Além das mutações únicas, os autores perguntam se o ESM‑2 consegue interpretar sequências inteiras de variantes conforme elas surgiram com o tempo. Eles incorporam uma sequência de spike para cada linhagem nomeada do SARS‑CoV‑2 e as posicionam em um mapa bidimensional usando uma abordagem chamada evo‑velocity, que também infere uma direção dominante de mudança. O layout resultante espelha a árvore filogenética conhecida: linhagens iniciais se agrupam, então ramos correspondentes a Alpha, Delta, Omicron e linhagens recombinantes se destacam na ordem temporal correta. Estatísticas sumárias simples, como gramaticalidade média e distância semântica, separam claramente linhagens não variantes, variantes iniciais de preocupação e vírus da classe Omicron, mostrando que a representação interna do modelo acompanha mudanças evolutivas significativas.
Transformando embeddings em um sistema de alerta precoce
Para explorar vigilância prática, a equipe introduz uma pontuação semântica dinâmica: cada nova sequência de spike é comparada não apenas à estirpe original de Wuhan, mas à média dos vírus circulantes nos três meses anteriores. Aplicada a dados de sequenciamento densos do Reino Unido, essa pontuação móvel produz ondas distintas à medida que Alpha, Delta e sublinhagens sucessivas de Omicron sobem e descem. Sequências que ficam a uma ou duas desvios-padrão da média atual são sinalizadas como potenciais sequências de preocupação. Usando apenas esses outliers iniciais, o método teria destacado a maioria das variantes de preocupação da Organização Mundial da Saúde e vários desdobramentos importantes posteriores, como JN.1, além de revelar quais sítios particulares na proteína spike são repetidamente alterados em linhagens emergentes.
O que isso significa para ameaças futuras
No geral, o estudo mostra que um modelo de linguagem de proteínas de uso geral, aplicado pronto para uso, pode identificar quais partes da proteína spike do SARS‑CoV‑2 são flexíveis, quais sítios são estruturalmente críticos, como as mutações interagem entre si e como a spike do vírus se deslocou pelo espaço evolutivo ao longo da pandemia. Como o método funciona a partir de uma única sequência proteica e não depende de alinhamentos pré‑existentes ou de dados estruturais detalhados, ele poderia ser aplicado muito cedo em um surto, quando apenas um punhado de genomas é conhecido. À medida que modelos semelhantes são refinados e ajustados a conjuntos de dados virais, eles podem se tornar parte importante do conjunto de ferramentas para prever como novos patógenos evoluem e para priorizar variantes para estudo laboratorial e desenho de vacinas.
Citação: Lamb, K.D., Hughes, J., Lytras, S. et al. From single-sequences to evolutionary trajectories: protein language models capture the evolutionary potential of SARS-CoV-2. Nat Commun 17, 2938 (2026). https://doi.org/10.1038/s41467-026-69569-9
Palavras-chave: modelos de linguagem de proteínas, spike do SARS-CoV-2, evolução viral, epistasia, vigilância de variantes