Clear Sky Science · es
De secuencias individuales a trayectorias evolutivas: los modelos de lenguaje proteico captan el potencial evolutivo del SARS‑CoV‑2
Por qué esto importa para futuras pandemias
Durante gran parte de la pandemia de COVID‑19, los científicos fueron a remolque: las nuevas variantes surgían en el mundo real antes de que los laboratorios pudieran medir qué implicaban esos cambios para la infectividad o el escape inmunitario. Este estudio demuestra que potentes modelos informáticos, diseñados originalmente para comprender el lenguaje humano, pueden leer en su lugar el “lenguaje” de las proteínas e inferir cómo es probable que cambie y se adapte la proteína espícula del coronavirus—usando solo su secuencia de bloques de construcción. Esa capacidad podría ayudar a los investigadores a detectar variantes preocupantes antes y puede generalizarse a muchos otros patógenos.
Enseñar a los ordenadores a leer proteínas
Los autores trabajan con un modelo de lenguaje proteico llamado ESM‑2, entrenado con decenas de millones de secuencias de proteínas procedentes de todo el árbol de la vida. De forma análoga a cómo un modelo de lenguaje aprende gramática y significado a partir de palabras, ESM‑2 aprende qué patrones de aminoácidos “tienen sentido” en proteínas reales. Al recibir la secuencia de la proteína espícula de SARS‑CoV‑2, el modelo asigna a cada posible mutación dos puntuaciones clave: una puntuación de gramaticalidad que refleja qué tan bien una secuencia cambiada encaja con las reglas aprendidas de la estructura proteica, y una puntuación semántica que mide cuánto difiere la proteína en la representación interna del modelo. Estas puntuaciones pueden calcularse para cada posible mutación simple en un ordenador, una estrategia conocida como escaneo profundo de mutaciones in silico. 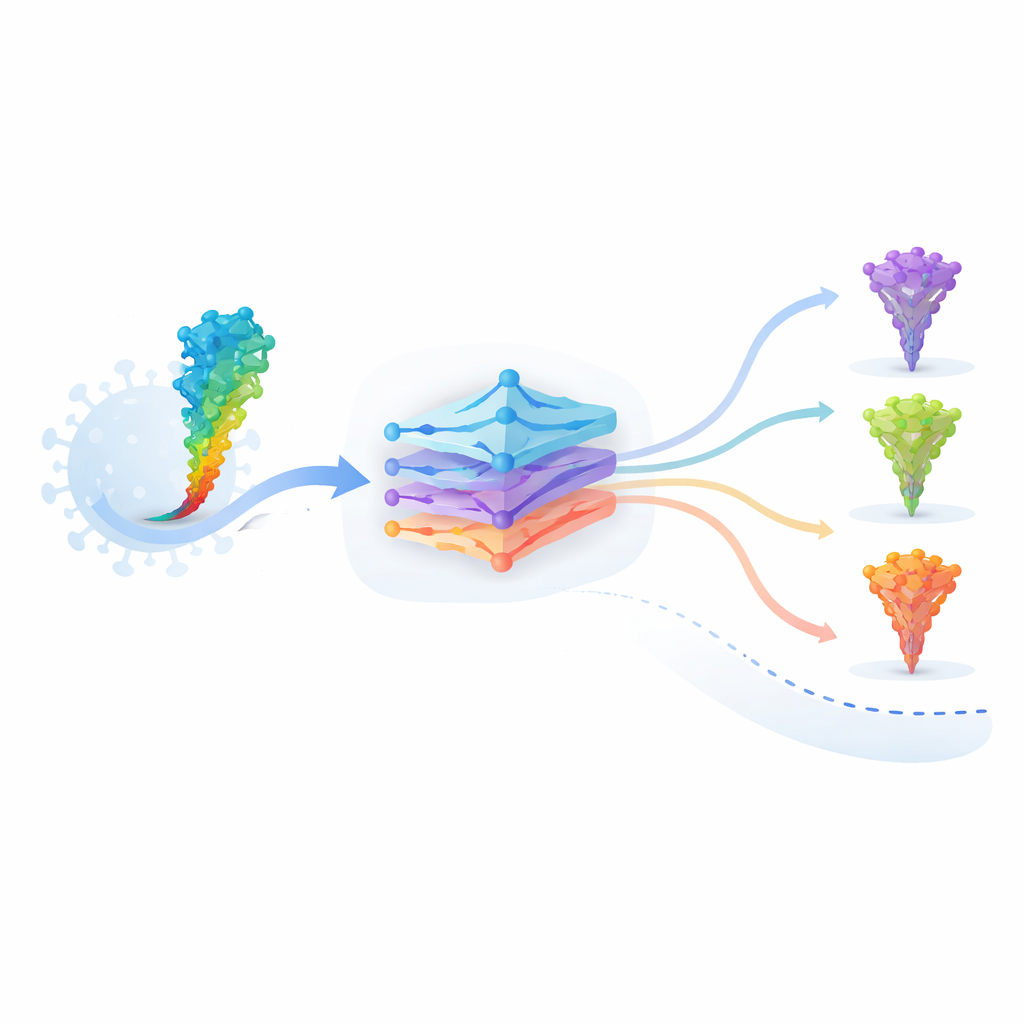
Mapear dónde el virus puede y no puede cambiar
Al examinar todos los cambios de una sola letra a lo largo de la espícula, el equipo encontró que ESM‑2 recupera de forma natural las principales características arquitectónicas de la proteína. La porción S2, que forma el tallo estable que impulsa la fusión de membranas, se predice como altamente constreñida: la mayoría de las mutaciones allí reducen drásticamente la gramaticalidad, lo que implica que dañarían la estructura o función de la proteína. En contraste, las regiones de la superficie externa de la porción S1, incluidos el dominio N‑terminal y el dominio de unión al receptor, toleran muchos más cambios. Esto coincide con lo observado en genomas virales reales, donde estas regiones expuestas acumulan mutaciones que ayudan al virus a unirse a las células y evadir anticuerpos, mientras que el núcleo estructural permanece más conservado.
Descubrir el trabajo en equipo oculto entre mutaciones
Las proteínas no son solo un conjunto de sitios independientes; una mutación puede cambiar cuán aceptables son otras, un fenómeno llamado epistasis. Los investigadores exploraron esto partiendo de la espícula de Omicron BA.1 y “revirtiendo” computacionalmente sus mutaciones definitorias, una a una, a la secuencia original de Wuhan. Cada reversión altera las probabilidades que el modelo asigna a aminoácidos en cada otra posición. Los grandes desplazamientos revelan pares de sitios cuyos destinos están vinculados. Con este enfoque, el estudio destaca puntos calientes conocidos como las posiciones alrededor de 484 y 501 en el dominio de unión al receptor, que en conjunto modulan tanto el escape inmunitario como la unión al receptor ACE2. También señala cúmulos menos obvios de residuos cuyas interacciones fueron confirmadas posteriormente en estudios experimentales sobre el crecimiento mejorado de Omicron en células nasales humanas, lo que sugiere que el modelo captura acoplamientos estructurales y funcionales genuinos. 
Seguir la evolución viral y detectar valores atípicos
Más allá de las mutaciones simples, los autores se preguntan si ESM‑2 puede dar sentido a secuencias completas de variantes tal como aparecieron a lo largo del tiempo. Insertan una secuencia de espícula por cada linaje nombrado de SARS‑CoV‑2 y las sitúan en un mapa bidimensional usando un enfoque llamado evo‑velocity, que además infiere una dirección dominante de cambio. La disposición resultante refleja el árbol filogenético conocido: los linajes tempranos se agrupan, luego se desprenden ramas correspondientes a Alpha, Delta, Omicron y linajes recombinantes en el orden temporal correcto. Estadísticos resumen simples, como la gramaticalidad media y la distancia semántica, separan claramente linajes no variantes, variantes tempranas de preocupación y virus de clase Omicron, mostrando que la representación interna del modelo sigue desplazamientos evolutivos significativos.
Convertir las incrustaciones en un sistema de alerta temprana
Para explorar la vigilancia práctica, el equipo introduce una puntuación semántica dinámica: cada nueva secuencia de espícula se compara no solo con la cepa original de Wuhan sino con el promedio de virus circulantes en los tres meses previos. Aplicada a datos de secuenciación densos del Reino Unido, esta puntuación móvil produce olas distintas a medida que Alpha, Delta y las sucesivas sublíneas de Omicron suben y bajan. Las secuencias que se sitúan a una o dos desviaciones estándar del promedio actual se etiquetan como posibles secuencias de interés. Usando solo estos primeros valores atípicos, el método habría destacado la mayoría de las variantes de preocupación de la Organización Mundial de la Salud y varios descendientes importantes posteriores como JN.1, además de revelar qué sitios particulares en la proteína espícula se alteran de forma recurrente en linajes emergentes.
Qué significa esto para amenazas futuras
En conjunto, el estudio demuestra que un modelo de lenguaje proteico de uso general, empleado tal cual, puede identificar qué partes de la proteína espícula de SARS‑CoV‑2 son flexibles, qué sitios son estructuralmente críticos, cómo interactúan las mutaciones y cómo la espícula del virus ha vagado por el espacio evolutivo a lo largo de la pandemia. Debido a que el método funciona a partir de una sola secuencia proteica y no depende de alineamientos preexistentes ni de datos estructurales detallados, podría aplicarse muy pronto en un brote, cuando solo se conocen unas pocas genomas. A medida que modelos similares se refinan y ajustan a conjuntos de datos virales, pueden convertirse en una parte importante de la caja de herramientas para prever cómo evolucionan nuevos patógenos y para priorizar variantes para estudios de laboratorio y diseño de vacunas.
Cita: Lamb, K.D., Hughes, J., Lytras, S. et al. From single-sequences to evolutionary trajectories: protein language models capture the evolutionary potential of SARS-CoV-2. Nat Commun 17, 2938 (2026). https://doi.org/10.1038/s41467-026-69569-9
Palabras clave: modelos de lenguaje proteico, espícula de SARS‑CoV‑2, evolución viral, epistasis, vigilancia de variantes