Clear Sky Science · de
Von Einzelsequenzen zu evolutionären Bahnen: Proteinsprachemodelle erfassen das evolutionäre Potenzial von SARS‑CoV‑2
Warum das für künftige Pandemien wichtig ist
Während der größten Teile der COVID‑19‑Pandemie liefen Wissenschaftlerinnen und Wissenschaftler hinterher: Neue Varianten tauchten in der realen Welt auf, bevor Labore messen konnten, was diese Veränderungen für Infektiosität oder Immunflucht bedeuteten. Diese Studie zeigt, dass leistungsfähige Computermodelle, ursprünglich entwickelt, um menschliche Sprache zu verstehen, stattdessen die „Sprache“ von Proteinen lesen und daraus ableiten können, wie sich das Spike‑Protein des Coronavirus wahrscheinlich verändern und anpassen wird — allein auf Grundlage seiner Bausteinsequenz. Diese Fähigkeit könnte Forschende dabei unterstützen, besorgniserregende Varianten früher zu erkennen und auf viele andere Erreger übertragbar sein.
Computern beibringen, Proteine zu lesen
Die Autorinnen und Autoren arbeiten mit einem Proteinsprachemodell namens ESM‑2, das an zig Millionen Proteinsequenzen aus dem gesamten Stammbaum des Lebens trainiert wurde. Ähnlich wie ein Sprachmodell Grammatik und Bedeutung aus Worten lernt, erkennt ESM‑2, welche Aminosäuremuster in echten Proteinen „Sinn machen“. Gibt man dem Modell die Sequenz des SARS‑CoV‑2‑Spikeproteins, vergibt es für jede mögliche Mutation zwei zentrale Werte: einen Grammatikalitätswert, der widerspiegelt, wie gut eine veränderte Sequenz zu den gelernten Regeln von Proteinstruktur passt, und einen Semantikwert, der misst, wie sehr sich das Gesamtprotein in der internen Repräsentation des Modells verändert. Diese Werte lassen sich für jede mögliche Einzelmutation am Computer berechnen — eine Strategie, die als in silico Deep Mutational Scanning bekannt ist. 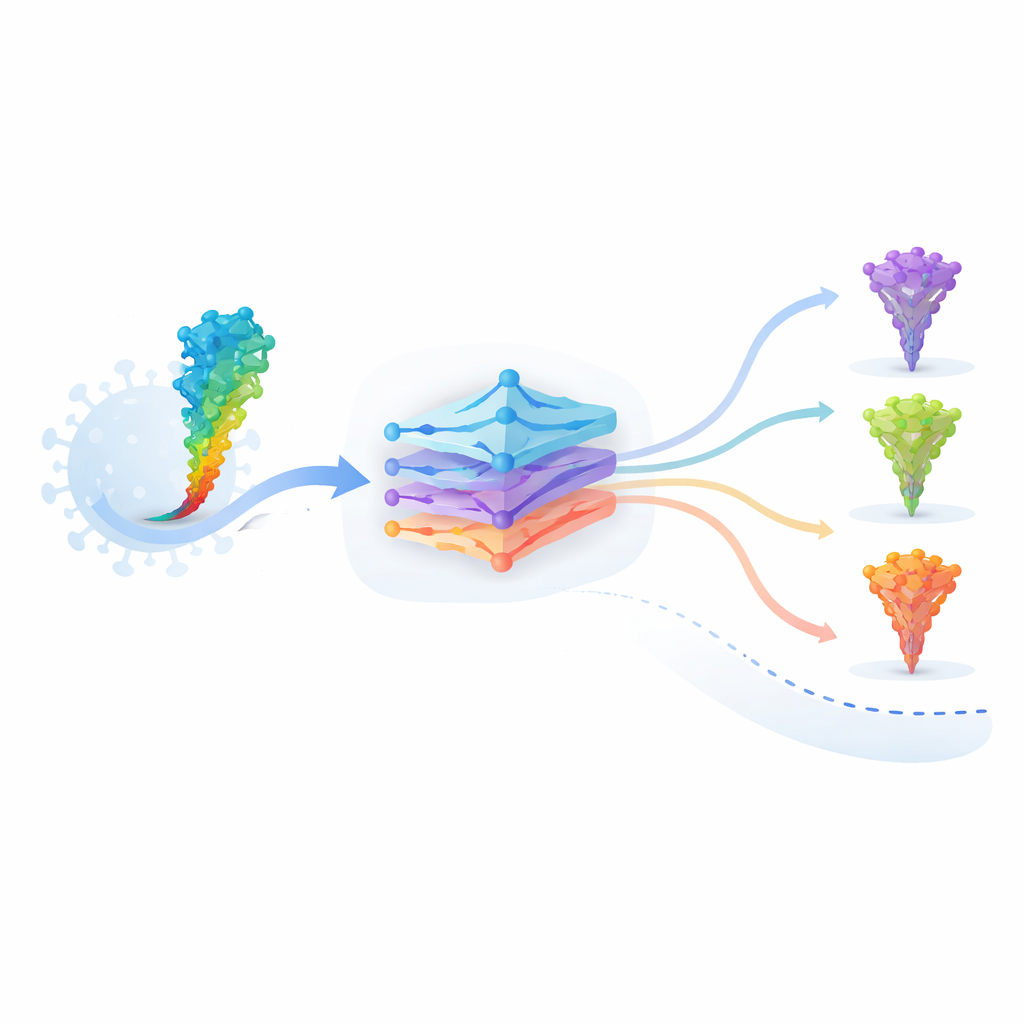
Aufzeigen, wo sich das Virus ändern kann und wo nicht
Durch das Scannen aller Einbuchstaben‑Änderungen über das Spike‑Protein hinweg stellte das Team fest, dass ESM‑2 die wesentlichen architektonischen Merkmale des Proteins von selbst rekonstruiert. Der S2‑Abschnitt, der den stabilen Stiel bildet, der die Membranfusion antreibt, wird als stark eingeschränkt vorhergesagt: Die meisten Mutationen dort verringern die Grammatikalität stark, was darauf hindeutet, dass sie die Proteinstruktur oder -funktion schädigen würden. Demgegenüber tolerieren Regionen auf der Außenseite des S1‑Teils, einschließlich der N‑terminalen Domäne und der Rezeptorbindungsdomäne, deutlich mehr Veränderungen. Das stimmt mit Beobachtungen in realen Virusgenomen überein, wo sich in diesen exponierten Bereichen Mutationen ansammeln, die dem Virus helfen, an Zellen anzudocken und Antikörpern zu entgehen, während der strukturelle Kern konservierter bleibt.
Verborgenes Zusammenspiel zwischen Mutationen aufdecken
Proteine sind nicht einfach eine Ansammlung unabhängiger Stellen; eine Mutation kann beeinflussen, wie verträglich andere Mutationen sind — ein Phänomen, das als Epistasis bezeichnet wird. Die Forschenden untersuchten dies, indem sie vom Omicron BA.1‑Spike ausgingen und dessen charakteristische Mutationen einzeln rechnerisch auf die ursprüngliche Wuhan‑Sequenz „zurücksetzten“. Jede Reversion verändert die Wahrscheinlichkeiten des Modells für Aminosäuren an allen anderen Positionen. Große Verschiebungen zeigen Paare von Stellen, deren Schicksal miteinander verknüpft ist. Mit diesem Ansatz hebt die Studie bekannte Hotspots hervor, wie Positionen um 484 und 501 in der Rezeptorbindungsdomäne, die gemeinsam sowohl die Immunflucht als auch die Bindung an den ACE2‑Rezeptor beeinflussen. Sie weist auch auf weniger offensichtliche Residuen‑Cluster hin, deren Wechselwirkungen später in experimentellen Studien zum verbesserten Wachstum von Omicron in menschlichen Nasenzellen bestätigt wurden — ein Hinweis darauf, dass das Modell echte strukturelle und funktionelle Kopplungen erfasst. 
Der viralen Evolution folgen und Ausreißer erkennen
Über Einzelmutationen hinaus fragten die Autorinnen und Autoren, ob ESM‑2 ganze Variantenfolgen, wie sie im Zeitverlauf auftauchten, sinnvoll einordnen kann. Sie betteten für jede benannte SARS‑CoV‑2‑Linie eine Spike‑Sequenz ein und positionierten diese in einer zweidimensionalen Karte mittels eines Ansatzes namens Evo‑Velocity, der auch eine dominierende Änderungsrichtung ableitet. Das resultierende Layout spiegelt den bekannten phylogenetischen Baum wider: Frühe Linien gruppieren sich zusammen, dann trennen sich Äste, die Alpha, Delta, Omicron und rekombinante Linien entsprechen, in der richtigen zeitlichen Reihenfolge ab. Einfache Zusammenfassungsstatistiken wie durchschnittliche Grammatikalität und semantische Distanz trennen sauber nicht‑variantige Linien, frühe besorgniserregende Varianten und Omicron‑artige Viren, was zeigt, dass die interne Repräsentation des Modells sinnvolle evolutionäre Verschiebungen nachverfolgt.
Einbettungen in ein Frühwarnsystem verwandeln
Um praktische Überwachung zu erforschen, führen die Forschenden einen dynamischen Semantik‑Score ein: Jede neue Spike‑Sequenz wird nicht nur mit dem ursprünglichen Wuhan‑Stamm verglichen, sondern mit dem Durchschnitt der in den vorangegangenen drei Monaten zirkulierenden Viren. Auf dichten Sequenzierungsdaten aus dem Vereinigten Königreich angewandt, erzeugt dieser gleitende Score deutlich voneinander getrennte Wellen, als Alpha, Delta und aufeinanderfolgende Omicron‑Sublinien auf‑ und absteigen. Sequenzen, die eine oder zwei Standardabweichungen vom aktuellen Durchschnitt entfernt liegen, werden als potenzielle Besorgnisfälle markiert. Hätte man nur diese frühen Ausreißer betrachtet, hätte die Methode die meisten von der Weltgesundheitsorganisation benannten Varianten von Besorgnis sowie mehrere wichtige spätere Abkömmlinge wie JN.1 hervorgehoben und gleichzeitig gezeigt, welche spezifischen Stellen im Spike‑Protein sich in aufkommenden Linien wiederholt verändern.
Was das für künftige Bedrohungen bedeutet
Insgesamt zeigt die Studie, dass ein allgemeines Proteinsprachemodell, direkt aus der Schachtel verwendet, identifizieren kann, welche Teile des SARS‑CoV‑2‑Spikeproteins flexibel sind, welche Stellen strukturell kritisch sind, wie Mutationen zusammenwirken und wie sich der Spike im Verlauf der Pandemie durch den evolutiven Raum bewegt hat. Da die Methode von einer einzelnen Proteinsequenz ausgeht und nicht auf vorbestehenden Alignments oder detaillierten Strukturdaten beruht, könnte sie sehr früh in einem Ausbruch angewendet werden, wenn nur eine Handvoll Genome bekannt sind. Wenn ähnliche Modelle weiter verfeinert und an virale Datensätze angepasst werden, könnten sie ein wichtiger Bestandteil des Werkzeugkastens zur Vorhersage der Entwicklung neuer Erreger sowie zur Priorisierung von Varianten für Laboruntersuchungen und Impfstoffdesign werden.
Zitation: Lamb, K.D., Hughes, J., Lytras, S. et al. From single-sequences to evolutionary trajectories: protein language models capture the evolutionary potential of SARS-CoV-2. Nat Commun 17, 2938 (2026). https://doi.org/10.1038/s41467-026-69569-9
Schlüsselwörter: Proteinsprachemodelle, SARS‑CoV‑2 Spike, virale Evolution, Epistasis, Variantenüberwachung