Clear Sky Science · ru
Улучшенное моделирование колебательной энергии диатомных молекул с использованием обобщённой дробной производной
Почему малые вибрации молекул имеют значение
Каждый ваш вдох, каждое пламя и каждое облако в космосе заполнены простыми двухатомными молекулами, которые постоянно вибрируют. Эти микроскопические колебания определяют, как молекулы поглощают свет, накапливают энергию и участвуют в химических реакциях. Чтобы понять и предсказать такое поведение, учёные строят математические модели движения молекул. В этой статье представлено новое средство более точного моделирования этих колебаний с помощью современного подхода к исчислению, применённого к старой, но мощной модели молекулы.
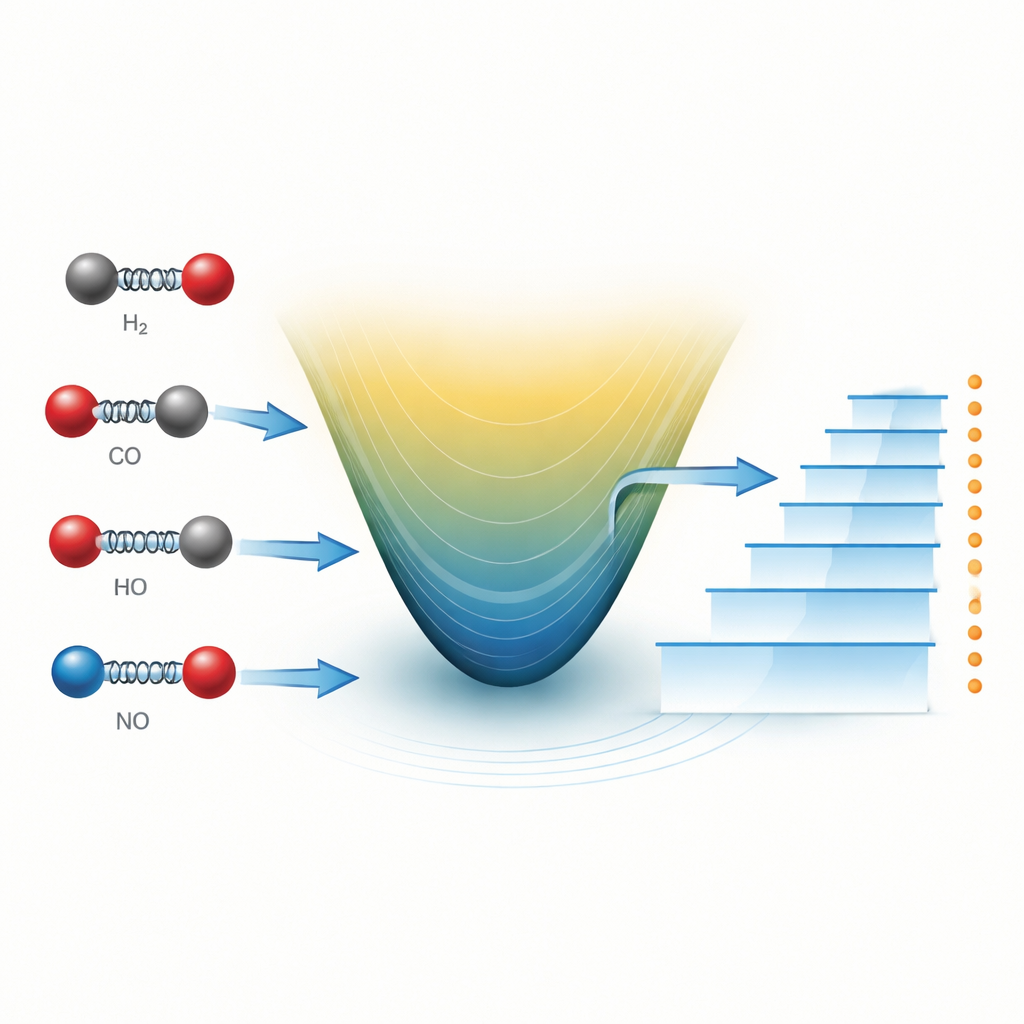
Как учёные обычно представляют дрожащую молекулу
Диатомную молекулу — состоящую всего из двух атомов — можно представить как две шарики, соединённые пружиной. Когда атомы приближаются или удаляются друг от друга, они перемещаются по энергетическому ландшафту вверх или вниз. Почти столетие простая формула, известная как потенциал Морса, служит рабочим инструментом для описания этого ландшафта. Она учитывает, как связь растягивается, уплотняется и в конечном счёте разрывается. В сочетании с уравнением Шрёдингера из квантовой механики потенциал Морса позволяет вычислять разрешённые уровни колебательной энергии, которые проявляются в виде резких линий в молекулярных спектрах, измеряемых в лаборатории или наблюдаемых в отдалённых звёздных средах.
Где классическая картина даёт сбои
Стандартная модель Морса лучше всего работает для молекул, которые не вращаются и не находятся в сильно возбуждённых состояниях. Однако реальные молекулы вращаются во время вибрации, и их поведение становится более сложным на высоких уровнях энергии. Чтобы учесть вращение, добавляют дополнительный энергетический барьер — так называемый центробежный член, — но это делает уравнения труднорешаемыми в точной форме. С течением времени были разработаны многочисленные хитрые приближения и численные приёмы, однако остаются небольшие расхождения между теорией и точными экспериментальными данными, особенно для высоких колебательных уровней и для молекул с различными электронными состояниями.
Новая дробная трактовка движения молекул
Авторы применяют недавнюю идею из «дробного» исчисления, которое обобщает обычные производные так, что их порядок может быть нецелым. В этой рамке способ отклика системы может нести память о прошлом либо эффективно кодировать тонкие, нелокальные взаимодействия. Опираясь на обобщённую дробную производную, сохраняющую ключевые правила стандартного исчисления, команда расширяет известный метод решения (метод Никифорова–Уварова) до того, что они называют обобщённым дробным методом НУ. С помощью этого подхода они получают аналитические формулы для уровней колебательной энергии молекул в любом числе пространственных измерений, начиная от потенциала Морса и обрабатывая центробежный барьер с помощью уточнённого приближения.
Проверка новой модели
Чтобы проверить, действительно ли дробный подход помогает, авторы применяют его к двадцати двум разным диатомным молекулам, включая виды, важные в химии, материаловедении и астрофизике, такие как CO, Na2, AlH, SiO+, TaO и TaS. Для каждого случая они строят кривые потенциальной энергии на основе известных молекулярных констант и сравнивают предсказанные колебательные уровни с высококачественными эталонными данными, полученными из анализов Ридберг–Клайн–Риз (RKR). Они рассматривают дробный порядок как небольшой подстраиваемый параметр, который эффективно суммирует сложные взаимодействия, не учтённые в более простой модели. Систематически минимизируя среднюю процентную ошибку для каждой молекулы, они показывают, что дробные формулы воспроизводят наблюдаемые колебательные энергии с замечательной точностью, часто улучшая согласие вдвое и более по сравнению с классическим пределом и успешно конкурируя с современными альтернативными потенциалами и численными методами.

Почему меньшие ошибки важны
Помимо средних мер ошибок, исследование изучает, как несоответствие между теорией и экспериментом меняется от уровня к уровню. Для многих молекул традиционная модель всё больше отклоняется от данных при увеличении колебательного квантового числа. В отличие от неё, дробная модель сохраняет эти отклонения менее выраженными и более однородными, особенно в верхней части спектра, где точные предсказания наиболее требовательны. Авторы также проверяют своё аналитическое решение для вращающихся состояний молекулы CO и находят отличное согласие с высокоточными численными эталонами, показывая, что их приближения остаются надёжными даже для заметного вращательного движения.
Что это означает для понимания молекул
Проще говоря, работа показывает, что мягкое «изгибание» привычных правил исчисления — разрешение производных нецелого порядка — ведёт к более гибкому и верному описанию того, как простые молекулы вибрируют и вращаются. Подбирая один дробный параметр, модель улавливает тонкие эффекты, которые ранее требовали более сложных потенциалов или тяжёлых численных расчётов. Это делает её мощным и эффективным инструментом для интерпретации молекулярных спектров в лаборатории, проектирования новых материалов и исследования состава астрономических сред, где изобилуют диатомные молекулы.
Цитирование: Khokha, E.M., Abu-Shady, M., Omugbe, E. et al. Improved modelling for vibrational energies of diatomic molecules using the generalized fractional derivative. Sci Rep 16, 12037 (2026). https://doi.org/10.1038/s41598-026-39091-5
Ключевые слова: диатомные молекулы, колебательные спектры, потенциал Морса, дробное исчисление, молекулярная спектроскопия