Clear Sky Science · nl
Verbeterde modellering van vibratie-energieën van diatomische moleculen met de gegeneraliseerde fractionele afgeleide
Waarom de kleine trillingen van moleculen ertoe doen
Elke ademhaling die je neemt, elke vlam die brandt en elke wolk in de ruimte bevat eenvoudige twee-atoommoleculen die constant trillen. Deze microscopische trillingen bepalen hoe moleculen licht absorberen, energie opslaan en deelnemen aan chemische reacties. Om dergelijk gedrag te begrijpen en te voorspellen bouwen wetenschappers wiskundige modellen van de beweging van moleculen. Dit artikel presenteert een nieuwe manier om die trillingen nauwkeuriger te modelleren, met een moderne draai aan calculus toegepast op een oud maar krachtig molecuulmodel.
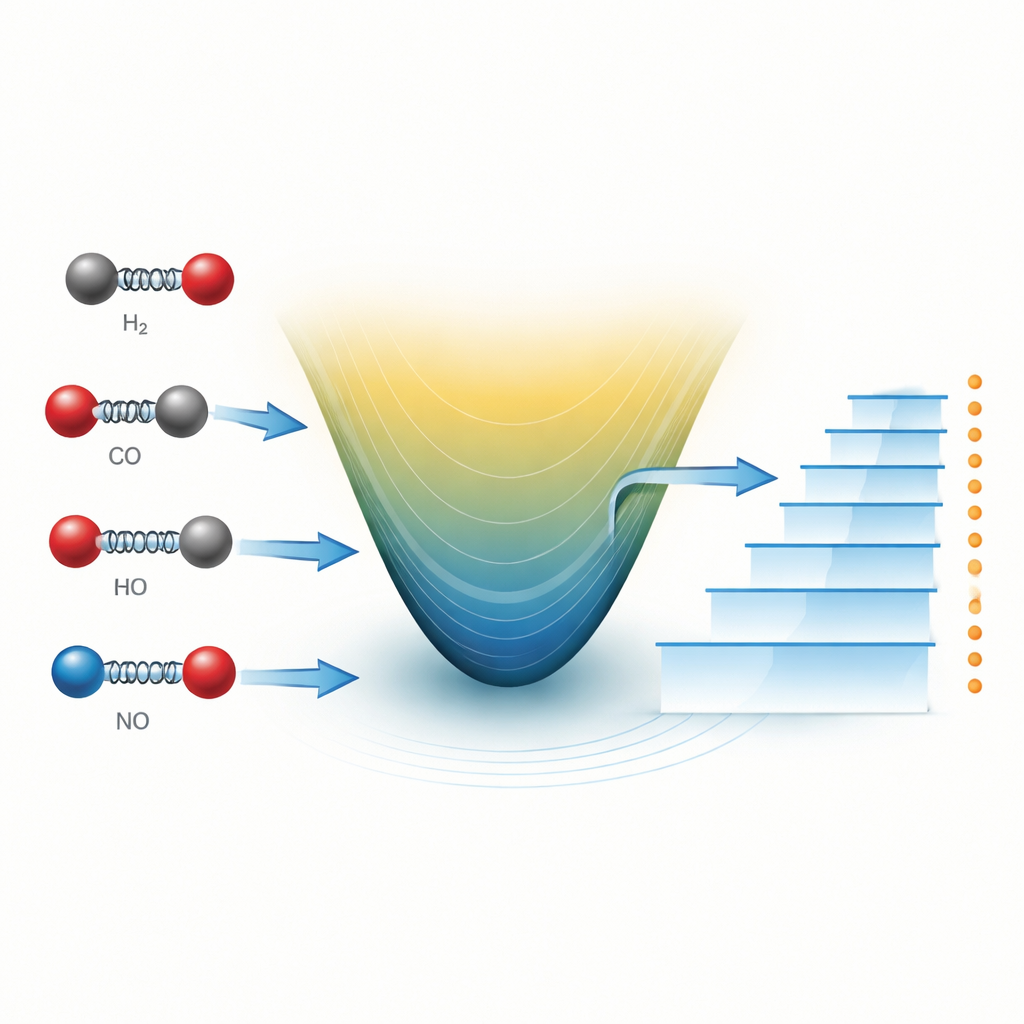
Hoe wetenschappers een trillend molecuul meestal voorstellen
Een diatomisch molecuul — bestaande uit slechts twee atomen — kan worden voorgesteld als twee balletjes verbonden door een veer. Wanneer de atomen dichter bij elkaar komen of verder uit elkaar bewegen, klimmen ze op of dalen ze af op een energielandschap. Al bijna een eeuw is een eenvoudige formule, bekend als de Morse-potentiaal, een werkpaard voor het beschrijven van dit landschap. Hij vangt hoe de binding uitrekt, verhardt en uiteindelijk breekt. Gecombineerd met de Schrödingervergelijking uit de kwantummechanica laat de Morse-potentiaal onderzoekers de toegestane vibratie-energieën berekenen, die verschijnen als scherpe lijnen in moleculaire spectra gemeten in het laboratorium of van verre sterren.
Waar het klassieke beeld tekortschiet
Het standaard Morse-model werkt het beste voor moleculen die niet draaien en niet te hoog geëxciteerd zijn. Reële moleculen draaien echter terwijl ze trillen, en hun gedrag wordt ingewikkelder bij hogere energieniveaus. Om rotatie te verwerken wordt een extra energiebijdrage — de centrifugale term — toegevoegd, maar dat maakt de vergelijkingen lastig exact op te lossen. Door de jaren heen zijn veel slimme benaderingen en numerieke trucs ontwikkeld, maar er blijven kleine afwijkingen bestaan tussen theorie en nauwkeurige experimentele gegevens, vooral voor hogere vibratieniveaus en voor moleculen met verschillende elektronische toestanden.
Een nieuwe fractionele twist op moleculaire beweging
De auteurs nemen een recent idee uit de "fractionele" calculus over, die gewone afgeleiden generaliseert zodat hun orde geen geheel getal hoeft te zijn. In dit kader kan de manier waarop een systeem reageert een geheugen van het verleden dragen, of effectief subtiele, niet-lokale interacties coderen. Uitgaand van een gegeneraliseerde fractionele afgeleide die sleutelregels van de standaardcalculus behoudt, breidt het team een bekende oplossingstechniek (de Nikiforov–Uvarov-methode) uit naar wat zij de gegeneraliseerde fractionele NU-methode noemen. Ze gebruiken deze aanpak om analytische formules te verkrijgen voor de vibratie-energieën van moleculen in een willekeurig aantal ruimtelijke dimensies, uitgaande van de Morse-potentiaal en waarbij de rotatiebarrière met een verfijnde benadering wordt behandeld.
Het nieuwe model op de proef stellen
Om te zien of de fractionele benadering echt helpt, passen de auteurs deze toe op tweeëntwintig verschillende diatomische moleculen, waaronder soorten die belangrijk zijn in de scheikunde, materiaalkunde en astrofysica zoals CO, Na2, AlH, SiO+, TaO en TaS. Voor elk geval construeren ze potentiële energiekrommen uit bekende molecuulconstanten en vergelijken de voorspelde vibratieniveaus met hoogwaardige referentiegegevens verkregen uit Rydberg–Klein–Rees (RKR)-analyses. Ze behandelen de fractionele orde als een kleine verstelbare parameter die effectief complexe interacties samenvat die het simpelere model niet captureert. Door systematisch de gemiddelde procentuele fout voor elk molecuul te minimaliseren, tonen ze aan dat de fractionele formules de waargenomen vibratie-energieën met opmerkelijke nauwkeurigheid reproduceren, vaak de overeenkomst met een factor twee of meer verbeteren ten opzichte van de klassieke limiet en succesvol concurreren met state-of-the-art alternatieve potentiaalmodellen en numerieke methoden.

Waarom kleinere fouten een groter verhaal vertellen
Buiten gemiddelde foutmaten onderzoekt de studie hoe de afwijking tussen theorie en experiment niveau per niveau verandert. Voor veel moleculen verwijdert het traditionele model zich verder van de gegevens naarmate het vibratiekwantumgetal toeneemt. In tegenstelling daarmee houdt het fractionele model die afwijkingen kleiner en gelijkmatiger, vooral in het bovenste deel van het spectrum waar nauwkeurige voorspellingen het meest veeleisend zijn. De auteurs testen ook hun analytische oplossing voor roterende toestanden van het CO-molecuul en vinden uitstekende overeenstemming met zeer precieze numerieke referentiedata, wat aantoont dat hun benaderingen betrouwbaar blijven zelfs bij aanzienlijke rotatiebeweging.
Wat dit betekent voor het begrip van moleculen
In alledaagse bewoordingen laat dit werk zien dat het voorzichtig "buigen" van de gebruikelijke regels van calculus — door afgeleiden van niet-gehele orde toe te staan — leidt tot een flexibeler en trouwere beschrijving van hoe eenvoudige moleculen trillen en roteren. Door één enkele fractionele parameter af te stemmen vangt het model subtiele effecten die voorheen complexere potentialen of zware numerieke berekeningen vereisten. Dat maakt het tot een krachtig en efficiënt hulpmiddel voor het interpreteren van moleculaire spectra in het laboratorium, het ontwerpen van nieuwe materialen en het onderzoeken van de samenstelling van astronomische omgevingen waar diatomische moleculen veel voorkomen.
Bronvermelding: Khokha, E.M., Abu-Shady, M., Omugbe, E. et al. Improved modelling for vibrational energies of diatomic molecules using the generalized fractional derivative. Sci Rep 16, 12037 (2026). https://doi.org/10.1038/s41598-026-39091-5
Trefwoorden: diatomische moleculen, vibratiespectra, Morse-potentiaal, fractionele calculus, moleculaire spectroscopie