Clear Sky Science · de
Verbesserte Modellierung von Schwingungsenergien diatomarer Moleküle unter Verwendung der verallgemeinerten fraktionalen Ableitung
Warum die winzigen Schwingungen von Molekülen wichtig sind
Jeder Atemzug, jede brennende Flamme und jede Wolke im All enthält einfache Zwei-Atom-Moleküle, die ständig vibrieren. Diese mikroskopischen Schwingungen bestimmen, wie Moleküle Licht absorbieren, Energie speichern und an chemischen Reaktionen teilnehmen. Um ein solches Verhalten zu verstehen und vorherzusagen, erstellen Wissenschaftler mathematische Modelle der Molekülbewegung. Dieser Artikel stellt eine neue Methode vor, diese Schwingungen genauer zu modellieren, indem eine moderne Variante der Analysis auf ein altes, aber leistungsfähiges Molekülmodell angewandt wird.
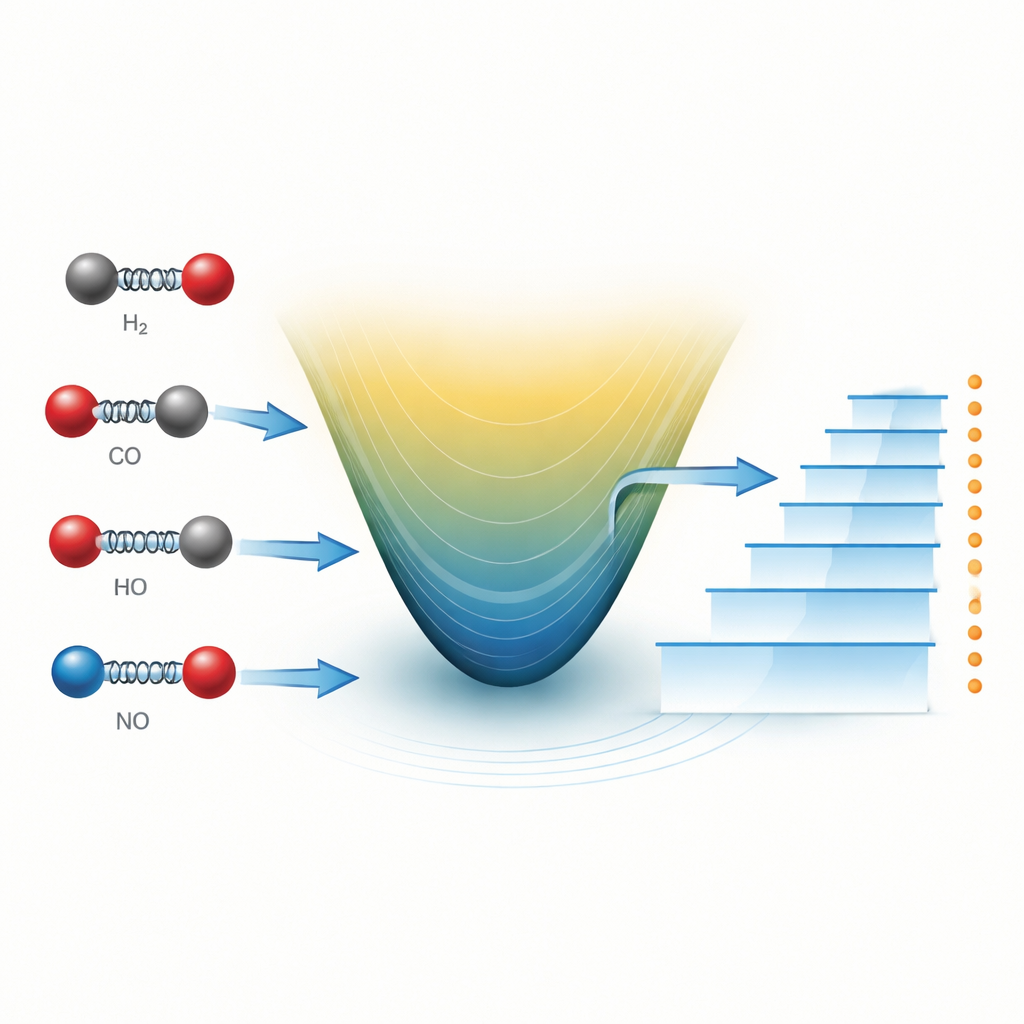
Wie Wissenschaftler sich ein schwingendes Molekül normalerweise vorstellen
Ein diatomares Molekül — bestehend aus nur zwei Atomen — kann man sich als zwei Kugeln vorstellen, die durch eine Feder verbunden sind. Wenn die Atome sich einander nähern oder sich weiter trennen, bewegen sie sich auf einer Energiefläche hinauf oder hinab. Seit fast einem Jahrhundert ist eine einfache Formel, bekannt als Morse-Potential, ein Arbeitspferd zur Beschreibung dieser Fläche. Sie erfasst, wie die Bindung sich dehnt, steifer wird und schließlich bricht. In Kombination mit der Schrödinger-Gleichung der Quantenmechanik erlaubt das Morse-Potential Forschern, die erlaubten Schwingungsenergien zu berechnen, die sich als scharfe Linien in molekularen Spektren zeigen — gemessen im Labor oder von fernen Sternen.
Worin das klassische Bild versagt
Das Standard-Morse-Modell funktioniert am besten für Moleküle, die sich nicht drehen und nicht zu stark angeregt sind. Reale Moleküle rotieren jedoch beim Schwingen, und ihr Verhalten wird bei höheren Energien komplexer. Um die Rotation zu berücksichtigen, wird ein zusätzlicher Energieanteil — der sogenannte Zentrifugalterm — hinzugefügt, was die Gleichungen jedoch schwer exakt lösbar macht. Im Laufe der Jahre wurden viele clevere Näherungen und numerische Tricks entwickelt, doch bleiben kleine Abweichungen zwischen Theorie und präzisen experimentellen Daten, insbesondere für höhere Schwingungsniveaus und für Moleküle in verschiedenen elektronischen Zuständen.
Eine neue fraktionale Wendung der Molekülbewegung
Die Autoren greifen eine jüngere Idee aus der „fraktionalen“ Analysis auf, die gewöhnliche Ableitungen verallgemeinert, sodass deren Ordnung nicht ganzzahlig sein kann. In diesem Rahmen kann das Ansprechverhalten eines Systems eine Erinnerung an seine Vergangenheit tragen oder effektiv subtile, nichtlokale Wechselwirkungen kodieren. Aufbauend auf einer verallgemeinerten fraktionalen Ableitung, die zentrale Regeln der gewöhnlichen Analysis bewahrt, erweitern die Forscher eine bekannte Lösungstechnik (die Nikiforov–Uvarov-Methode) zu dem, was sie die verallgemeinerte fraktionale NU-Methode nennen. Mit diesem Ansatz gewinnen sie analytische Formeln für die Schwingungsenergien von Molekülen in beliebiger Raumdimension, ausgehend vom Morse-Potential und unter Behandlung der Rotationsbarriere mit einer verfeinerten Näherung.
Das neue Modell wird auf die Probe gestellt
Um zu prüfen, ob der fraktionale Ansatz tatsächlich hilft, wenden die Autoren ihn auf zweiundzwanzig verschiedene diatomare Moleküle an, darunter Spezies von Bedeutung in Chemie, Materialwissenschaft und Astrophysik wie CO, Na2, AlH, SiO+, TaO und TaS. Für jeden Fall konstruieren sie Potentialenergiekurven aus bekannten molekularen Konstanten und vergleichen die vorhergesagten Schwingungsniveaus mit hochwertigen Referenzdaten aus Rydberg–Klein–Rees-(RKR-)Analysen. Sie behandeln die fraktionale Ordnung als kleinen einstellbaren Parameter, der komplexe Wechselwirkungen effektiv zusammenfasst, die das einfachere Modell nicht erfasst. Durch systematisches Minimieren des durchschnittlichen Prozentfehlers für jedes Molekül zeigen sie, dass die fraktionalen Formeln die beobachteten Schwingungsenergien mit bemerkenswerter Genauigkeit reproduzieren und oft die Übereinstimmung um den Faktor zwei oder mehr gegenüber dem klassischen Grenzfall verbessern, wobei sie erfolgreich mit modernen alternativen Potentialen und numerischen Methoden konkurrieren.

Warum kleinere Fehler eine größere Aussagekraft haben
Über durchschnittliche Fehlermessungen hinaus untersucht die Studie, wie die Abweichung zwischen Theorie und Experiment von Niveau zu Niveau variiert. Für viele Moleküle driftet das traditionelle Modell mit wachsender Schwingungsquantenzahl weiter von den Daten weg. Im Gegensatz dazu hält das fraktionale Modell diese Abweichungen kleiner und gleichmäßiger, besonders im oberen Bereich des Spektrums, wo genaue Vorhersagen am anspruchsvollsten sind. Die Autoren testen außerdem ihre analytische Lösung für rotierende Zustände des CO-Moleküls und finden eine ausgezeichnete Übereinstimmung mit sehr präzisen numerischen Referenzdaten, was zeigt, dass ihre Näherungen auch bei beträchtlicher Rotation zuverlässig bleiben.
Was das für das Verständnis von Molekülen bedeutet
Alltäglich ausgedrückt zeigt die Arbeit, dass das behutsame „Biegen" der üblichen Regeln der Analysis — durch Zulassen von Ableitungen nichtganzzahliger Ordnung — zu einer flexibleren und treueren Beschreibung führt, wie einfache Moleküle schwingen und rotieren. Durch das Einstellen eines einzigen fraktionalen Parameters erfasst das Modell subtile Effekte, die zuvor kompliziertere Potentiale oder aufwändige numerische Rechnungen erforderten. Das macht es zu einem leistungsfähigen und effizienten Werkzeug zur Interpretation molekularer Spektren im Labor, zum Entwurf neuer Materialien und zum Analysieren der Zusammensetzung astronomischer Umgebungen, in denen diatomare Moleküle häufig vorkommen.
Zitation: Khokha, E.M., Abu-Shady, M., Omugbe, E. et al. Improved modelling for vibrational energies of diatomic molecules using the generalized fractional derivative. Sci Rep 16, 12037 (2026). https://doi.org/10.1038/s41598-026-39091-5
Schlüsselwörter: diatomare Moleküle, Schwingungsspektren, Morse-Potential, fraktionale Analysis, molekulare Spektroskopie