Clear Sky Science · pt
Inclinação octaédrica e deslocamento do sítio B em perovskitas halogenadas não são acoplados
Por que as vibrações do cristal importam para a tecnologia do futuro
Perovskitas halometálicas são candidatas promissoras para células solares e termelétricos porque seus átomos e elétrons são incomumente móveis. Esse movimento constante determina quão bem elas absorvem luz, transportam carga e conduzem calor. Neste estudo, os pesquisadores fazem uma pergunta simples, porém crucial: quando os blocos construtores desses cristais se inclinam e quando seus átomos centrais se deslocam para fora do centro, esses movimentos estão ligados ou agem de forma independente? A resposta muda a forma como os cientistas pensam em ajustar esses materiais para tecnologias de energia mais limpa.
Dois tipos de movimento dentro de uma gaiola cristalina
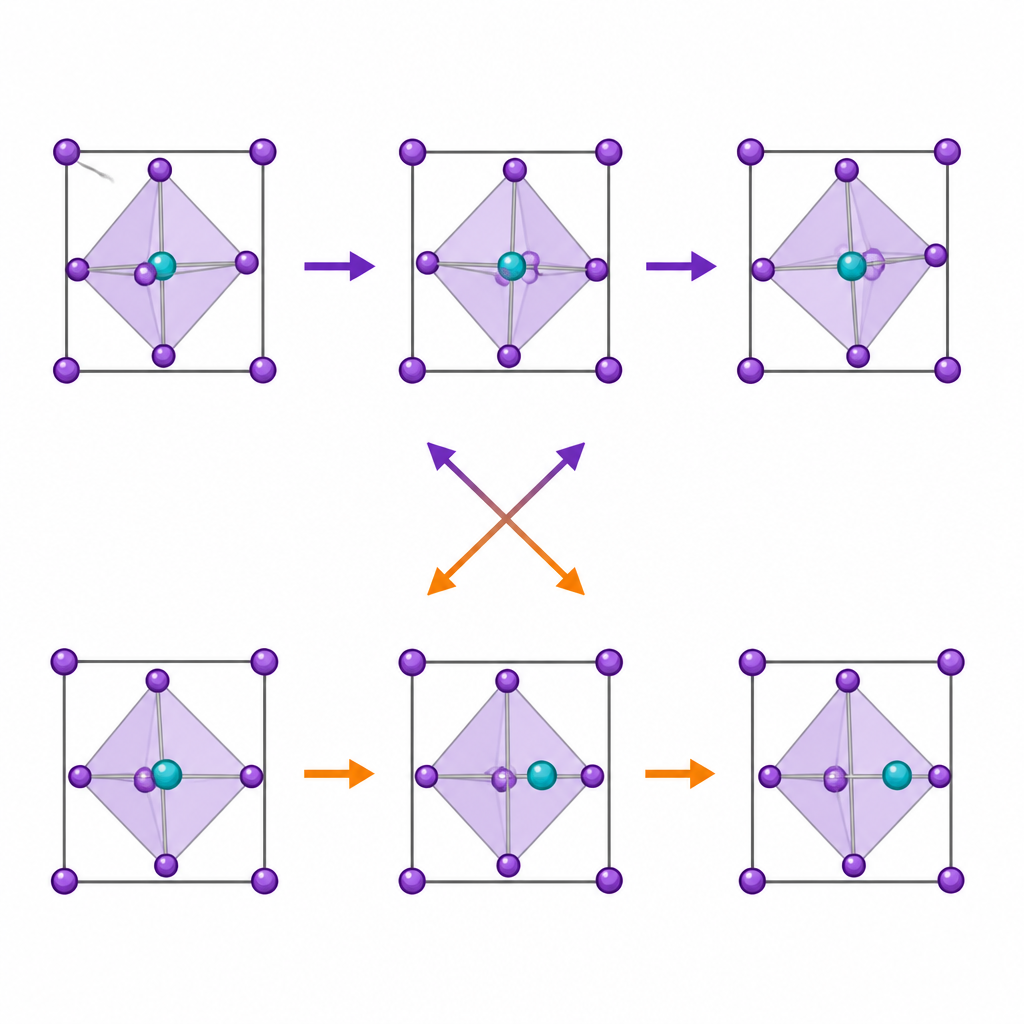
Perovskitas são construídas a partir de gaiolas repetitivas em forma de octaedro: um átomo metálico central cercado por seis íons halogenados. Essas gaiolas não ficam perfeitamente estáticas. Um movimento chave é a inclinação octaédrica, em que gaiolas vizinhas giram em direções opostas, alterando sutilmente os ângulos entre os átomos. Outro é o deslocamento do centro (off-centering), no qual o átomo metálico central desliza para fora do meio de sua gaiola, impulsionado por uma nuvem eletrônica assimétrica, conhecida como par isolado. Ambos os movimentos influenciam como os elétrons se movimentam e como o material responde à luz e ao calor, por isso muitos pesquisadores presumiam que eles estavam ligados.
Seguindo os elétrons enquanto eles se deslocam e oscilam
Para sondar a ligação entre esses movimentos, os autores simularam três cristais intimamente relacionados, todos com a mesma estrutura geral: césio, bromo e um metal no centro que é chumbo, estanho ou germânio. Esses metais apresentam pares isolados com força crescente de chumbo para estanho até germânio. Usando dinâmica molecular de primeiros princípios, eles acompanharam tanto as posições atômicas quanto as nuvens eletrônicas ao longo do tempo em temperatura elevada. Em seguida, analisaram a simetria dessas flutuações com ferramentas matemáticas que funcionam como impressões digitais para diferentes padrões de movimento, permitindo separar com precisão inclinação e deslocamento do centro.
Deslocando centros sem impulsionar inclinações
As simulações revelam que, à medida que o par isolado se torna mais pronunciado, o metal central desloca-se mais para fora do centro, especialmente no caso do germânio. Contudo, a quantidade de inclinação octaédrica diminui ao longo da mesma série. Testes estatísticos cuidadosos mostram que o grau de deslocamento do centro e a força do par isolado não têm, essencialmente, correlação direta com o movimento de inclinação. As duas distorções ocupam canais de simetria diferentes dentro do cristal, o que significa que não se fundem em um único modo compartilhado. Em vez de se ajudarem, competem: quando o deslocamento do centro é forte, a inclinação é suprimida, e quando a inclinação é facilitada, o deslocamento do centro é mais modesto.
Papel oculto da força da ligação química
Se o par isolado não dirige diretamente a inclinação, o que a controla? A chave está em quão fortemente o metal e os átomos de bromo compartilham elétrons. À medida que o átomo central muda de chumbo para estanho e para germânio, a ligação entre metal e bromo torna-se mais direcional e parcialmente covalente. A densidade eletrônica no bromo aponta com mais clareza para o metal, enrijecendo a estrutura octaédrica. Isso torna mais difícil as gaiolas rotacionarem, mesmo enquanto o mesmo par isolado incentiva o metal a deslocar-se do centro. Análises temporais dos movimentos confirmam esse quadro: em cristais à base de germânio, o metal deslocado se move de forma mais lenta entre posições enquanto as vibrações de inclinação são relativamente rígidas e rápidas; em cristais à base de chumbo, ambos os movimentos são mais suaves e flexíveis.
Alavancas de projeto para materiais melhores
Como inclinação e deslocamento do centro não estão travados um ao outro, projetistas de materiais podem, em princípio, ajustá-los separadamente. Ajustar a força das ligações por escolha química, pressão ou tensão pode enrijecer ou amolecer os movimentos de inclinação sem necessariamente desligar os deslocamentos polares do átomo central. Isso importa porque a inclinação remodela caminhos eletrônicos e o fluxo de calor, enquanto o deslocamento do centro afeta o comportamento dielétrico e os campos elétricos locais. O estudo mostra que controlar a simetria eletrônica e o caráter das ligações oferece uma rota para engenhar perovskitas onde a carga viaja com eficiência, o calor é gerenciado de forma eficaz e as mudanças estruturais podem ser direcionadas para funções desejadas em células solares, emissores de luz e dispositivos termelétricos.
Citação: Hylton-Farrington, C.M., Remsing, R.C. Octahedral tilting and B-site off-centering in halide perovskites are not coupled. Nat Commun 17, 4345 (2026). https://doi.org/10.1038/s41467-026-70882-6
Palavras-chave: perovskitas halogenadas, inclinação octaédrica, par de elétrons isolado, deslocamento do centro, rigidez da ligação