Clear Sky Science · it
L'inclinazione degli ottaedri e lo spostamento dal centro del sito B nei perovskiti alogenuri non sono accoppiati
Perché i sobbalzi del cristallo contano per le tecnologie future
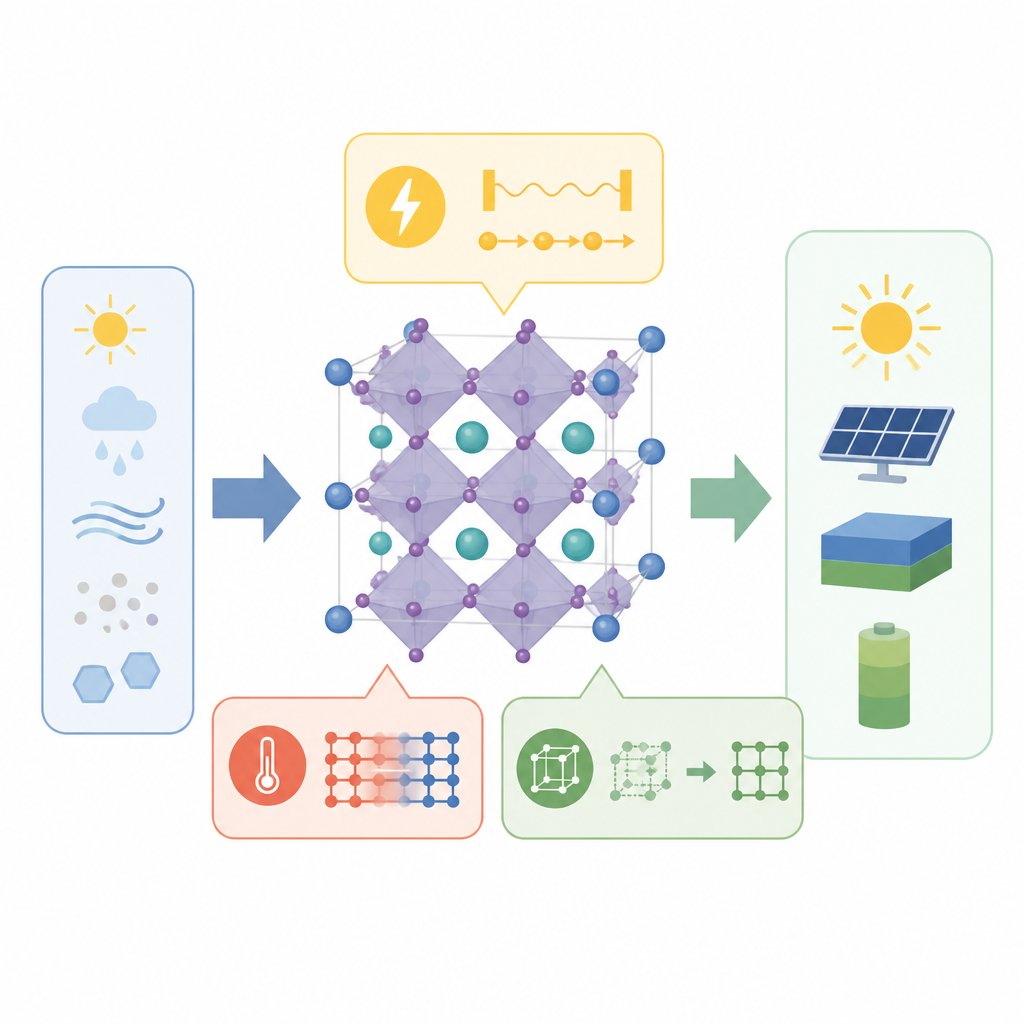
I perovskiti alogenuri metallici sono stelle emergenti per celle solari e termoelettrici perché i loro atomi ed elettroni sono insolitamente mobili. Questo moto incessante influenza il modo in cui assorbono la luce, trasportano carica e conducono calore. In questo studio i ricercatori pongono una domanda semplice ma cruciale: quando i piccoli blocchi costitutivi di questi cristalli si inclinano e quando gli atomi centrali si spostano dal centro, questi moti sono collegati oppure agiscono indipendentemente? La risposta cambia il modo in cui gli scienziati pensano di ottimizzare questi materiali per tecnologie energetiche più pulite.
Due tipi di moto dentro una gabbia cristallina
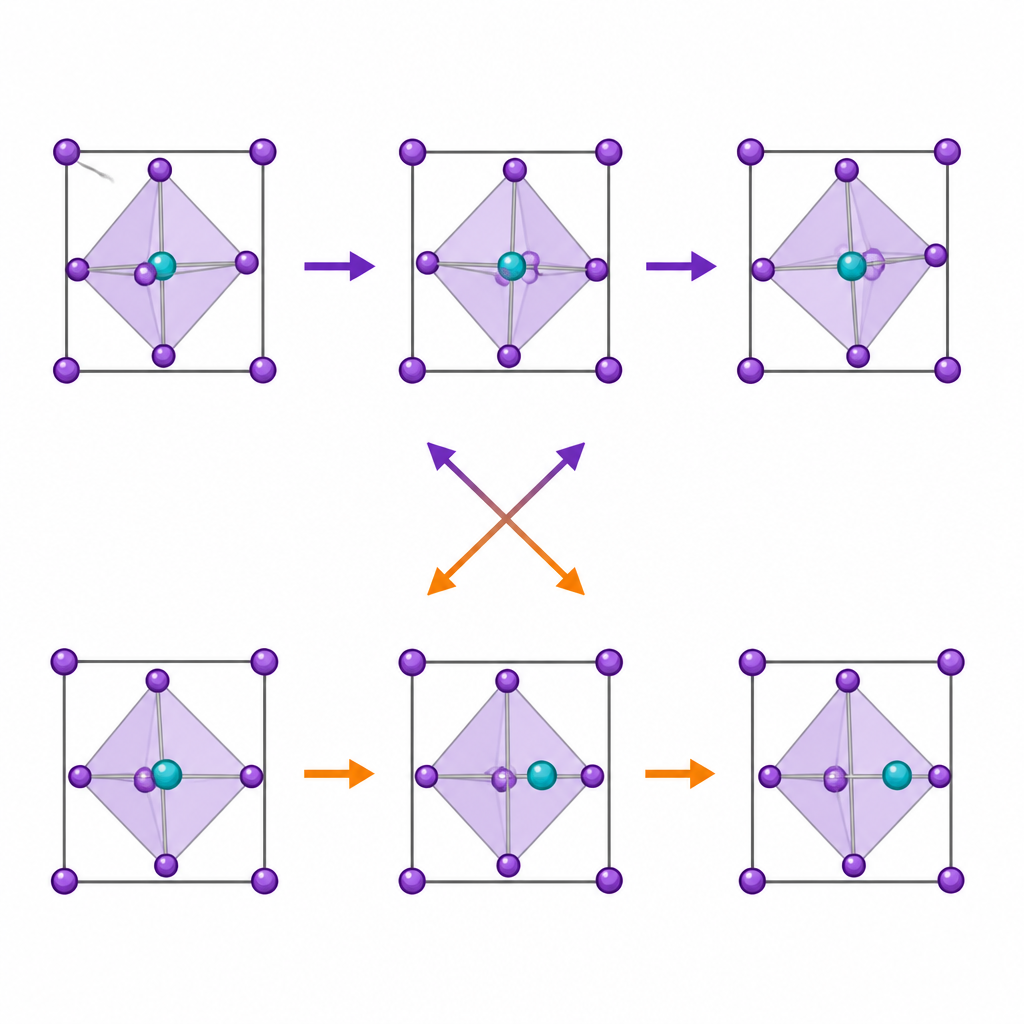
I perovskiti sono costituiti da gabbie ripetute a forma di ottaedro: un atomo metallico centrale circondato da sei ioni alogeni. Queste gabbie non stanno perfettamente ferme. Un moto chiave è l'inclinazione ottaedrica, in cui gabbie vicine ruotano in direzioni opposte, modificando sottilmente gli angoli tra gli atomi. Un altro è lo spostamento dal centro, in cui l'atomo metallico centrale scivola fuori dal centro della sua gabbia, guidato da una nube elettronica asimmetrica, nota come coppia solitaria. Entrambi i moti influenzano come gli elettroni si muovono e come il materiale risponde a luce e calore, perciò molti ricercatori avevano supposto che fossero collegati.
Seguire gli elettroni mentre si spostano e oscillano
Per sondare il legame tra questi moti, gli autori hanno simulato tre cristalli strettamente correlati, tutti con la stessa struttura complessiva: cesio, bromo e un metallo al centro che è rispettivamente piombo, stagno o germanio. Questi metalli portano coppie solitarie di forza crescente da piombo a stagno a germanio. Usando dinamica molecolare da primi principi, hanno seguito sia le posizioni atomiche sia le nubi elettroniche nel tempo a temperatura elevata. Hanno quindi analizzato la simmetria di queste fluttuazioni con strumenti matematici che funzionano come impronte digitali per diversi schemi di moto, permettendo di separare in modo preciso inclinazioni e spostamenti dal centro.
Spostare i centri senza far scattare le inclinazioni
Le simulazioni rivelano che man mano che la coppia solitaria diventa più pronunciata, il metallo centrale si sposta sempre più dal centro, soprattutto per il germanio. Tuttavia, la quantità di inclinazione ottaedrica diminuisce in realtà lungo la stessa serie. Test statistici accurati mostrano che il grado di spostamento dal centro e l'intensità della coppia solitaria non hanno essenzialmente alcuna correlazione diretta con il moto di inclinazione. Le due distorsioni occupano canali di simmetria diversi all'interno del cristallo, il che significa che non si fondono in un unico modo condiviso. Invece di favorirsi a vicenda, competono: quando lo spostamento dal centro è forte, l'inclinazione è soppressa, e quando l'inclinazione è agevole, lo spostamento dal centro è più modesto.
Ruolo nascosto della forza del legame chimico
Se la coppia solitaria non guida direttamente l'inclinazione, cosa lo fa? La chiave risiede in quanto fortemente l'atomo metallico e il bromo condividono elettroni. Cambiando l'atomo centrale da piombo a stagno a germanio, il legame tra metallo e bromo diventa più diretto e parzialmente covalente. La densità elettronica sul bromo punta più chiaramente verso il metallo, irrigidendo il telaio ottaedrico. Questo rende più difficile la rotazione delle gabbie, pur mentre la stessa coppia solitaria incoraggia il metallo a spostarsi dal centro. Analisi temporali dei moti confermano questo quadro: nei cristalli a base di germanio, il metallo spostato si muove lentamente tra posizioni mentre le vibrazioni di inclinazione sono relativamente strette e veloci; nei cristalli a base di piombo, entrambi i moti sono più morbidi e flessibili.
Manopole di progetto per materiali migliori
Poiché inclinazione e spostamento dal centro non sono vincolati tra loro, i progettisti di materiali possono, in principio, sintonizzarli separatamente. Regolare la forza del legame tramite scelta chimica, pressione o deformazione può irrigidire o ammorbidire i moti di inclinazione senza necessariamente disattivare gli spostamenti polari dell'atomo centrale. Questo è importante perché l'inclinazione rimodella i percorsi elettronici e il flusso di calore, mentre lo spostamento dal centro influenza il comportamento dielettrico e i campi elettrici locali. Lo studio mostra che controllare la simmetria elettronica e il carattere del legame offre una via per ingegnerizzare perovskiti in cui la carica si muove efficacemente, il calore è gestito in modo efficiente e i cambiamenti strutturali possono essere indirizzati verso funzioni desiderate in celle solari, emettitori di luce e dispositivi termoelettrici.
Citazione: Hylton-Farrington, C.M., Remsing, R.C. Octahedral tilting and B-site off-centering in halide perovskites are not coupled. Nat Commun 17, 4345 (2026). https://doi.org/10.1038/s41467-026-70882-6
Parole chiave: perovskiti alogenuri, inclinazione ottaedrica, coppia solitaria, spostamento dal centro, rigidità del legame