Clear Sky Science · de
Oktaedrische Kippung und B‑Stellen‑Verschiebung in Halogenperowskiten sind nicht gekoppelt
Warum Kristall‑Wackeln für zukünftige Technik wichtig ist
Metallhalogen‑Perowskite sind vielversprechend für Solarzellen und Thermoelektrik, weil ihre Atome und Elektronen ungewöhnlich mobil sind. Diese rastlose Bewegung bestimmt, wie gut sie Licht absorbieren, Ladung transportieren und Wärme leiten. In dieser Studie stellen die Forschenden eine einfache, aber entscheidende Frage: Wenn die winzigen Bausteine dieser Kristalle kippen und wenn ihre Zentralatome aus der Mitte rutschen, sind diese Bewegungen miteinander verbunden oder verhalten sie sich unabhängig? Die Antwort verändert, wie Wissenschaftler darüber nachdenken, diese Materialien für saubere Energietechnologien zu optimieren.
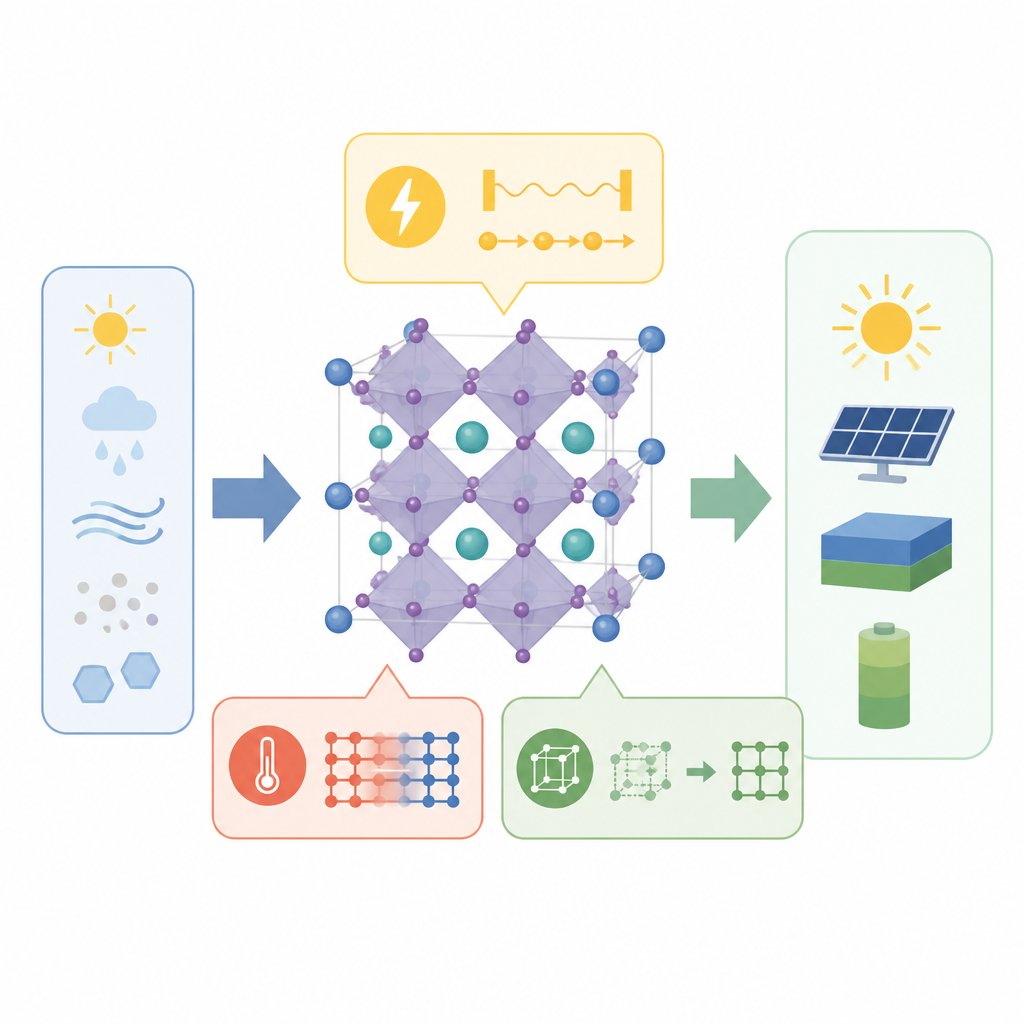
Zwei Arten von Bewegung im Kristallkäfig
Perowskite bestehen aus sich wiederholenden Käfigen in Form von Oktaedern: ein zentrales Metallatom umgeben von sechs Halogenid‑Ionen. Diese Käfige stehen nicht völlig still. Eine wesentliche Bewegung ist die oktaedrische Kippung, bei der benachbarte Käfige entgegengesetzt rotieren und so die Winkel zwischen den Atomen leicht verändern. Eine andere ist die Verschiebung aus der Mitte, bei der das zentrale Metallatom weg vom Mittelpunkt seines Käfigs gleitet, angetrieben durch eine ungleichmäßige Elektronenwolke, das sogenannte einsame Elektronenpaar. Beide Bewegungen beeinflussen, wie Elektronen sich bewegen und wie das Material auf Licht und Wärme reagiert, weshalb viele Forschende annahmen, sie stünden miteinander in Zusammenhang.
Elektronen folgen, während sie sich verschieben und schwanken
Um die Verbindung zwischen diesen Bewegungen zu untersuchen, simulierten die Autorinnen und Autoren drei eng verwandte Kristalle, alle mit derselben Gesamtstruktur: Cäsium, Brom und ein zentrales Metall, das entweder Blei, Zinn oder Germanium ist. Diese Metalle tragen einsame Elektronenpaare mit zunehmender Stärke von Blei über Zinn zu Germanium. Mit erstprinzipiellen Molekulardynamik‑Simulationen verfolgten sie über die Zeit sowohl die atomaren Positionen als auch die Elektronenwolken bei hoher Temperatur. Anschließend analysierten sie die Symmetrien dieser Fluktuationen mit mathematischen Werkzeugen, die wie Fingerabdrücke für verschiedene Bewegungsmuster wirken, und konnten so Kippung und Verschiebung präzise trennen.
Verschobene Zentren ohne Kippungsantrieb
Die Simulationen zeigen, dass mit stärkerem einsamen Elektronenpaar das zentrale Metall stärker aus der Mitte verschoben wird, besonders bei Germanium. Gleichzeitig nimmt jedoch die Stärke der oktaedrischen Kippung entlang derselben Reihe tatsächlich ab. Sorgfältige statistische Tests zeigen, dass das Ausmaß der Verschiebung und die Stärke des einsamen Elektronenpaars praktisch keine direkte Korrelation mit der Kippung aufweisen. Die beiden Verzerrungen besetzen unterschiedliche Symmetriekanäle im Kristall, das heißt, sie verschmelzen nicht zu einem gemeinsamen Modus. Statt sich gegenseitig zu verstärken, konkurrieren sie: Wenn die Verschiebung stark ist, wird die Kippung unterdrückt, und wenn Kippung leicht möglich ist, bleibt die Verschiebung eher moderat.
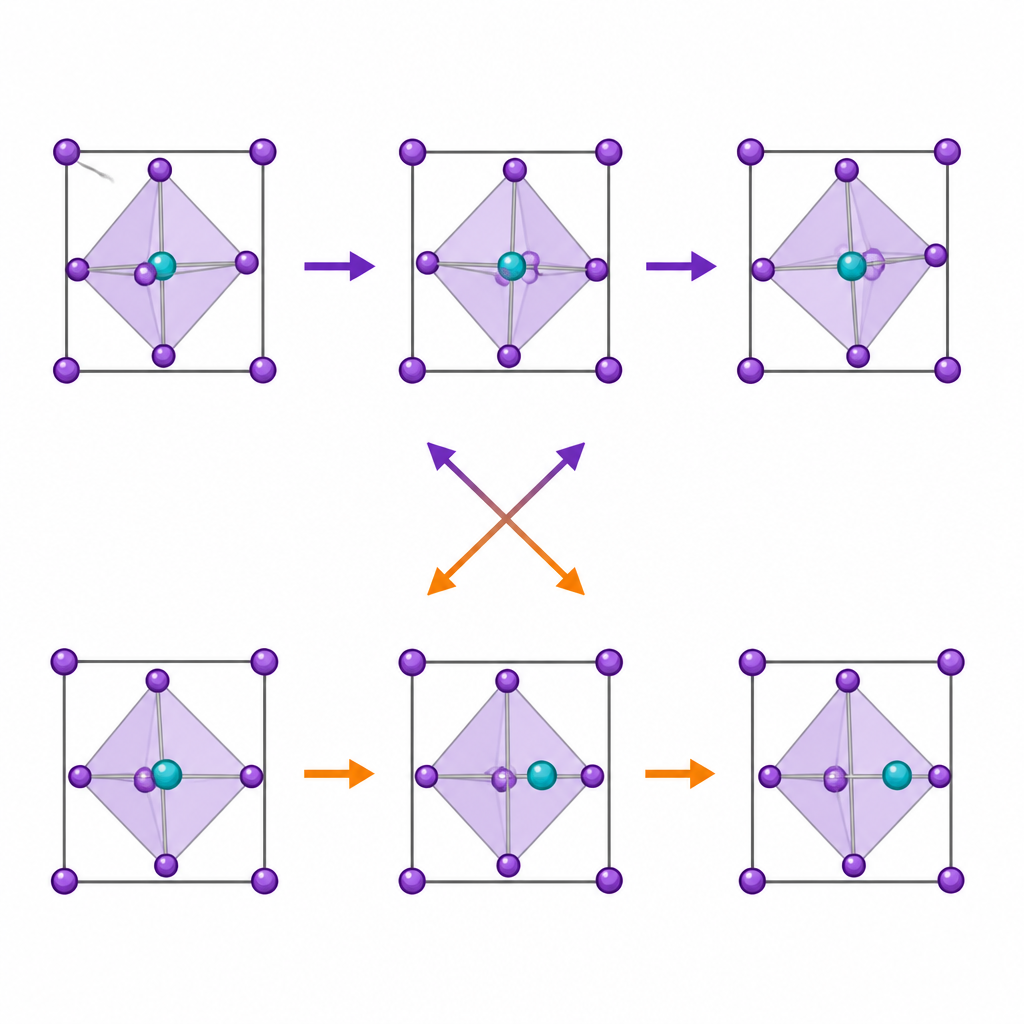
Versteckte Rolle der chemischen Bindungsstärke
Wenn das einsame Elektronenpaar die Kippung nicht direkt antreibt, was dann? Der Schlüssel liegt in der Frage, wie stark Metall und Brom Elektronen teilen. Wenn das zentrale Atom von Blei zu Zinn zu Germanium wechselt, wird die Bindung zwischen Metall und Brom richtungsbezogener und teilweise kovalenter. Die Elektronendichte am Brom weist klarer zum Metall, wodurch das oktaedrische Gerüst versteift wird. Das erschwert das Rotieren der Käfige, selbst während das gleiche einsame Elektronenpaar das Metall zum Aus‑der‑Mitte‑Bewegen ermuntert. Zeitaufgelöste Analysen der Bewegungen bestätigen dieses Bild: In Germanium‑basierten Kristallen bewegt sich das aus der Mitte geratene Metall träg zwischen Positionen, während Kippungsschwingungen relativ eng und schnell sind; in Blei‑basierten Kristallen sind beide Bewegungen weicher und flexibler.
Regelknöpfe für bessere Materialien
Weil Kippung und Verschiebung nicht fest miteinander gekoppelt sind, können Materialdesigner sie prinzipiell getrennt steuern. Durch Anpassung der Bindungsstärke mittels chemischer Auswahl, Druck oder Dehnung lässt sich die Kippungssteifigkeit erhöhen oder verringern, ohne notwendigerweise die polaren Verschiebungen des Zentralatoms auszuschalten. Das ist wichtig, weil Kippung die elektronischen Pfade und den Wärmetransport umgestaltet, während Verschiebung das dielektrische Verhalten und lokale elektrische Felder beeinflusst. Die Studie zeigt, dass die Kontrolle über elektronische Symmetrie und Bindungscharakter einen Weg bietet, Perowskite so zu entwerfen, dass Ladung effizient transportiert wird, Wärme gut gehandhabt wird und strukturelle Veränderungen für gewünschte Funktionen in Solarzellen, Lichtemittern und thermoelektrischen Bauelementen steuerbar sind.
Zitation: Hylton-Farrington, C.M., Remsing, R.C. Octahedral tilting and B-site off-centering in halide perovskites are not coupled. Nat Commun 17, 4345 (2026). https://doi.org/10.1038/s41467-026-70882-6
Schlüsselwörter: Halogenperowskite, oktaedrische Kippung, einsames Elektronenpaar, Verschiebung von der Mitte, Bindungssteifigkeit