Clear Sky Science · pt
Acoplamento cruzado de Stille de glicídeos de próxima geração ativado por ligante para a síntese estereoespecífica de aril C-glicósidos estéricamente congestionados
Por que é difícil construir medicamentos ligados a açúcares
Muitos medicamentos modernos imitam os açúcares complexos que revestem a superfície das nossas células. Quando um fragmento aromático planar de um fármaco é ligado diretamente a um açúcar por uma ligação carbono–carbono robusta, o resultado — um aril C-glicósido — pode ser muito mais estável no organismo do que uma ligação de açúcar normal. Essas estruturas aparecem em antibióticos, agentes anticâncer e novos medicamentos contra diabetes, mas são notoriamente difíceis de sintetizar, especialmente quando o anel ligado é volumoso e congestionado. Este artigo descreve uma nova forma de construir tais conexões açúcar–anel “estéricamente impedidas” de maneira limpa e confiável, abrindo caminho para um desenho mais rápido de medicamentos à base de açúcares.
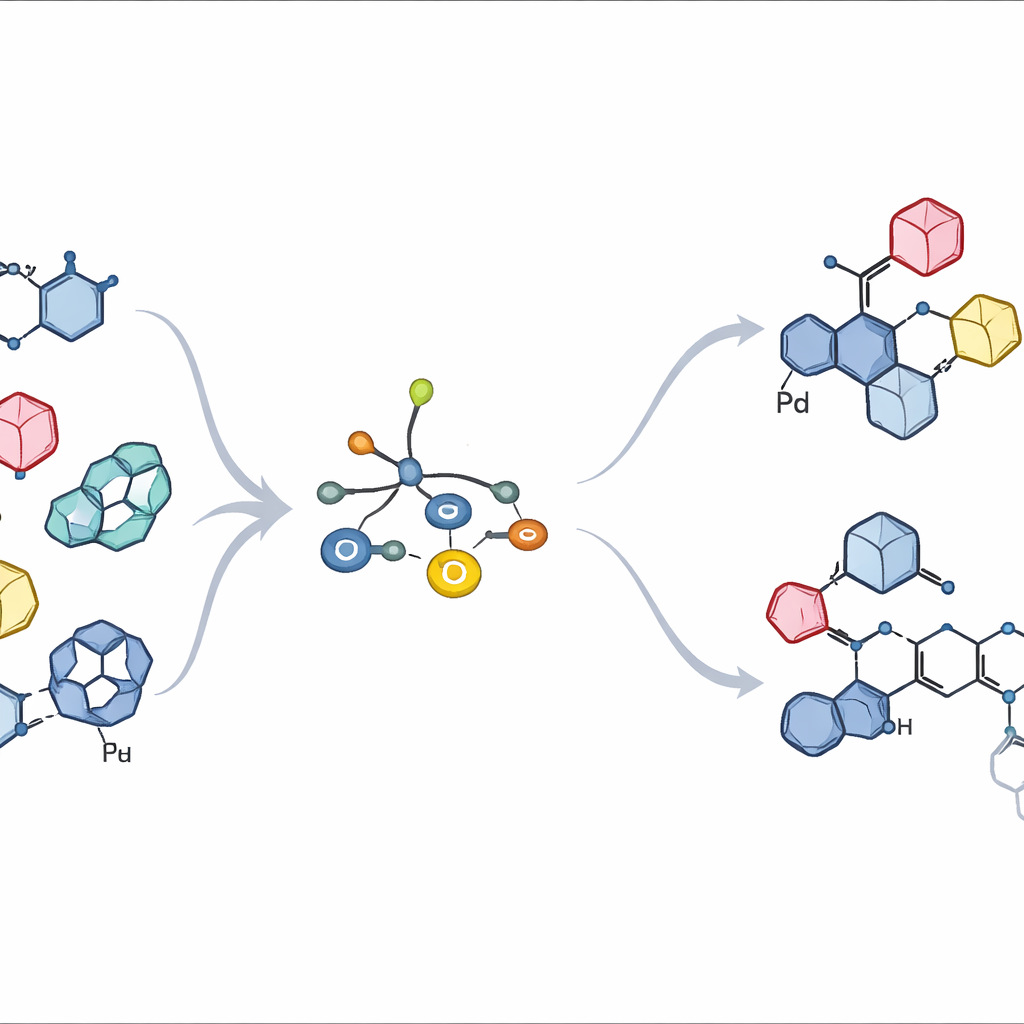
Ligação de açúcar teimosa em uma junção congestionada
Em açúcares naturais, o ponto chave de ligação é um único carbono na ponta do anel, conhecido como centro anomérico. Os químicos devem controlar não apenas qual parceiro se liga ali, mas também se a nova ligação aponta “para cima” ou “para baixo” no espaço — duas formas que podem se comportar como moléculas inteiramente diferentes em biologia. Métodos tradicionais frequentemente passam por fragmentos de açúcar altamente reativos carregados positivamente ou por intermediários radicalares. Essas rotas podem favorecer apenas uma direção, embaralhar a geometria inicial ou gerar uma mistura de subprodutos indesejados, especialmente quando o anel parceiro é grande, rígido e substituído por outros grupos. Como resultado, muitos aril C-glicósidos valiosos permanecem fora de alcance ou exigem sínteses longas e geradoras de desperdício.
Desenhando um ajudante metálico mais inteligente
Os autores partem de uma ideia mais recente: usar um açúcar contendo estanho que transfere seu carbono diretamente para um catalisador de paládio em um chamado acoplamento cruzado de Stille. Em trabalhos anteriores, uma família de ligantes fosfina ao redor do paládio permitiu que açúcares com estanho se acoplassem a anéis aromáticos simples mantendo perfeitamente a orientação original para cima ou para baixo. Contudo, quando o parceiro aromático se tornava volumoso — como os encontrados em fármacos reais — o mesmo sistema desacelerava ou parava. Aqui, a equipe redesenhou sistematicamente a estrutura do ligante, ajustando tanto seu volume quanto seu poder retirador de elétrons. Dois novos ligantes, chamados L10 e L11, surgiram como vencedores claros. Eles apresentam grupos arílicos fluorados fortemente atraentes ligados ao fósforo e braços contendo oxigênio cuidadosamente posicionados nos anéis de suporte, que em conjunto tornam o paládio mais propenso a aceitar o açúcar sem impedir a liberação do produto final.
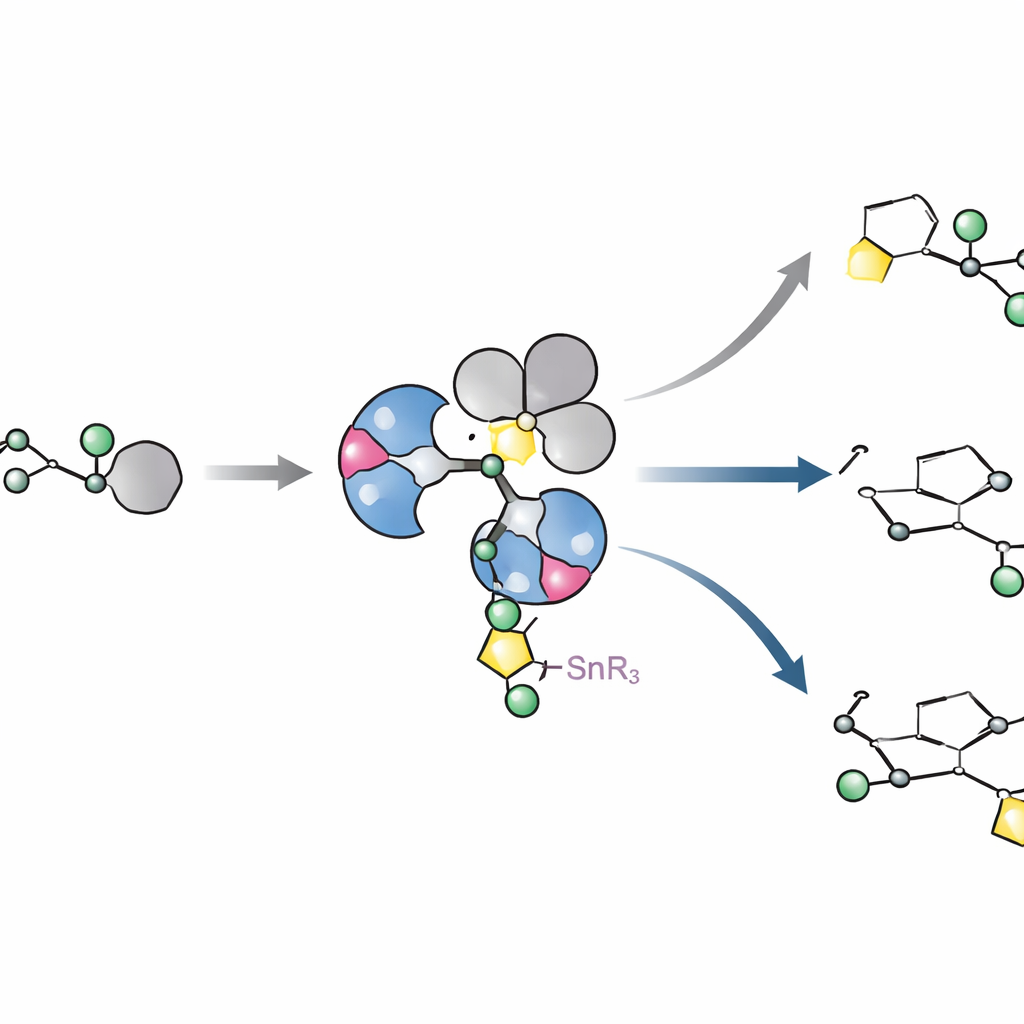
Vendo como o catalisador realmente é e se comporta
Para entender por que esses ligantes funcionam tão bem, os autores isolaram e cristalizaram complexos chave de paládio formados com haletos arílicos volumosos. Surpreendentemente, em vez das simples estruturas de metal único que se supunha havia muito tempo, eles observaram consistentemente dímeros: dois centros de paládio pontes por íons haleto. Esse arranjo provou ser uma característica natural de toda essa classe de ligantes, não uma exceção rara. Experimentos cinéticos e cálculos quântico-químicos então traçaram como esse dímero se separa o suficiente para reagir com o açúcar contendo estanho, transfere o fragmento de açúcar em um único passo que preserva a geometria e finalmente forma a desejada ligação carbono–carbono. As simulações mostraram que L11 desloca sutilmente a densidade eletrônica de modo que a formação da ligação é preferida em relação a uma via concorrente que poderia arrancar um grupo metoxi e desperdiçar material.
De reações modelo a alvos com evidente semelhança a fármacos
Munidos dessas percepções, a equipe testou uma ampla gama de parceiros aromáticos congestionados e doadores de açúcar. Mais de 65 exemplos — incluindo açúcares protegidos, desoxi-açúcares e até cadeias curtas de açúcares — foram ligados em rendimentos bons a excelentes com controle essencialmente perfeito sobre se a ligação final apontava para cima ou para baixo. Notavelmente, até açúcares completamente não protegidos, que possuem múltiplos grupos hidroxila livres, puderam ser acoplados de forma limpa, algo que normalmente causa reações secundárias severas. O método também funcionou na modificação em estágio avançado de fármacos aprovados e moléculas bioativas: ao instalar primeiro um único átomo de halogênio no anel aromático de um medicamento e então aplicar o novo acoplamento, os autores conectaram unidades de açúcar para criar novos análogos em apenas um ou dois passos.
Novos atalhos para híbridos complexos açúcar–fármaco
Para demonstrar o poder da abordagem, os autores simplificaram a síntese do medicamento para diabetes Enavogliflozina e variantes relacionadas, formando diretamente sua congestionada ligação açúcar–arila em uma etapa e acessando ambas as formas anoméricas possíveis. Eles também prepararam sistemas de anéis fundidos nos quais o açúcar e a unidade aromática compartilham um novo anel, estruturas anteriormente acessíveis apenas por rotas mais longas e delicadas. Comparações com métodos antigos baseados em cátions e radicais destacaram que essas abordagens ou falham completamente ou perdem o controle anomérico com os parceiros mais congestionados, enquanto o novo protocolo mantém a precisão.
O que isso significa para futuros medicamentos
Em termos cotidianos, este trabalho entrega um “conector de encaixe” confiável para unir açúcares complexos a fragmentos de fármacos volumosos em uma orientação tridimensional escolhida, mesmo em locais extremamente congestionados. Ao revelar que as espécies ativas de paládio são dímeros pontes por haletos e mostrar como ajustes sutis no ligante direcionam a reação longe de caminhos laterais desperdiçadores, o estudo também oferece regras gerais de projeto para futuros catalisadores. Juntos, esses avanços ampliam a caixa de ferramentas dos químicos que buscam fármacos modificados por açúcares mais estáveis e seletivos e devem acelerar a descoberta de antibióticos de próxima geração, agentes anticâncer e tratamentos para doenças metabólicas.
Citação: Yang, B., Chen, S., Han, Y. et al. Ligand-enabled next-generation glycosyl Stille cross-coupling for the stereospecific synthesis of sterically hindered aryl C-glycosides. Nat Commun 17, 3015 (2026). https://doi.org/10.1038/s41467-026-69859-2
Palavras-chave: aril C-glicósidos, acoplamento cruzado de glicídeos, catálise por paládio, projeto de ligantes, fármacos glicomiméticos