Clear Sky Science · en
Ligand-enabled next-generation glycosyl Stille cross-coupling for the stereospecific synthesis of sterically hindered aryl C-glycosides
Why building sugar-linked medicines is hard
Many modern medicines mimic the complex sugars that decorate the surface of our cells. When a flat ring-shaped fragment from a drug is joined directly to a sugar by a sturdy carbon–carbon bond, the result—an aryl C-glycoside—can be far more stable in the body than a normal sugar link. These structures appear in antibiotics, anticancer agents, and new diabetes drugs, but they are notoriously difficult to make, especially when the attached ring is bulky and crowded. This paper describes a new way to build such “sterically hindered” sugar–ring connections cleanly and reliably, opening doors to faster design of sugar-based medicines.
Stubborn sugar bonds at a crowded junction
In natural sugars, the key attachment point is a single carbon at the tip of the ring, known as the anomeric center. Chemists must control not only which partner attaches there, but also whether the new bond points “up” or “down” in space—two forms that can behave like entirely different molecules in biology. Traditional methods often pass through highly reactive positively charged sugar fragments or radical intermediates. These routes can favor only one direction, scramble the starting geometry, or create a mix of unwanted side products, especially when the partner ring is large, stiff, and decorated with other groups. As a result, many valuable aryl C-glycosides remain out of reach or require long, wasteful syntheses.
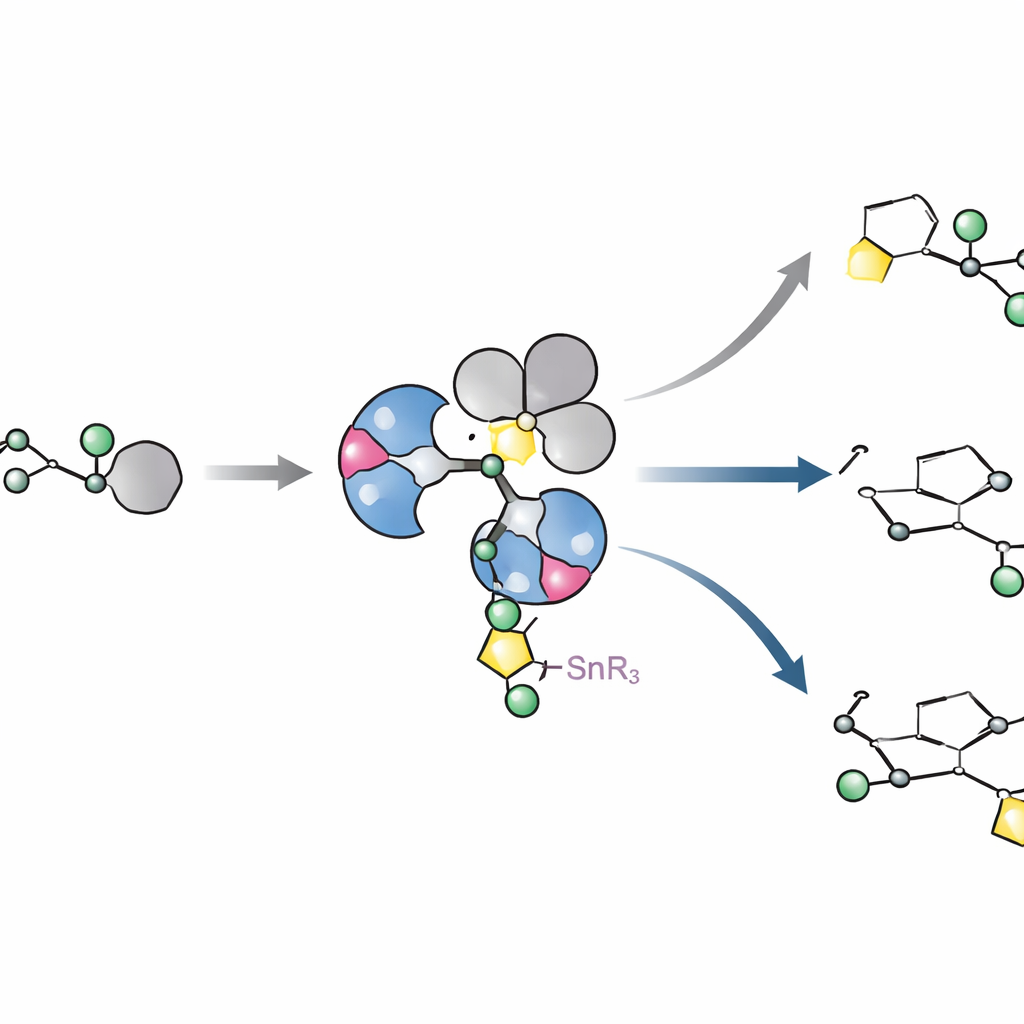
Designing a smarter metal helper
The authors build on a more recent idea: use a tin-bearing sugar that hands its carbon directly to a palladium catalyst in a so-called Stille cross-coupling. In earlier work, a family of phosphine ligands surrounding palladium allowed sugars with tin to couple with simple aromatic rings while preserving the original up or down orientation perfectly. However, when the aromatic partner became bulky—like those found in real drugs—the same system slowed down or stopped. Here, the team systematically redesigned the ligand framework, fine-tuning both its bulk and its electron-withdrawing power. Two new ligands, called L10 and L11, emerged as clear winners. They feature strongly pulling fluorinated aryl groups bound to phosphorus and carefully placed oxygen-containing arms on the supporting aryl rings, which together make palladium more eager to accept the sugar while still able to release the final product.
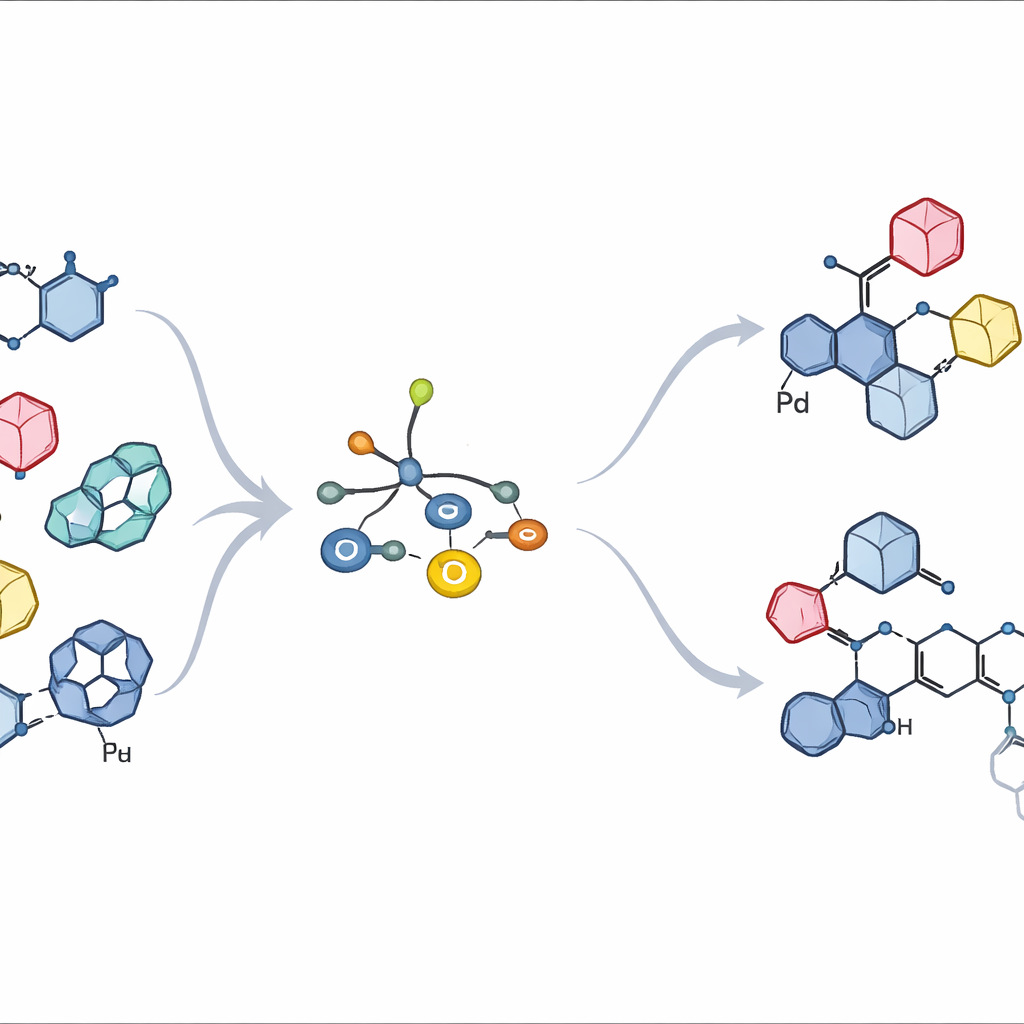
Seeing how the catalyst really looks and behaves
To understand why these ligands work so well, the authors isolated and crystallized key palladium complexes formed with bulky aryl halides. Surprisingly, instead of the simple single-metal structures that had long been assumed, they consistently observed dimers: two palladium centers bridged by halide ions. This arrangement proved to be a natural feature of this entire ligand class, not a rare exception. Kinetic experiments and quantum-chemical calculations then traced how this dimer breaks apart just enough to react with the tin-bearing sugar, transfers the sugar fragment in a single, geometry-preserving step, and finally forms the desired carbon–carbon bond. The computations showed that L11 subtly shifts electron density so that bond formation is preferred over a competing pathway that would chop off a methoxy group and waste material.
From model reactions to real drug-like targets
Armed with these insights, the team tested a wide range of crowded aromatic partners and sugar donors. More than 65 examples—including protected sugars, deoxy sugars, and even short sugar chains—were joined in good to excellent yields with essentially perfect control over whether the final bond pointed up or down. Remarkably, even completely unprotected sugars, which bear multiple free hydroxyl groups, could be coupled cleanly, something that usually causes severe side reactions. The method also worked in the “late-stage” modification of approved drugs and bioactive molecules: by first installing a single halogen atom on a drug’s aromatic ring and then applying the new coupling, the authors snapped on sugar units to make new analogues in just one or two steps.
New shortcuts to complex sugar–drug hybrids
To showcase the power of the approach, the authors streamlined the synthesis of the diabetes drug Enavogliflozin and related variants, directly forging its congested sugar–aryl bond in one step and accessing both possible anomeric forms. They also prepared fused ring systems in which the sugar and aromatic unit share a new ring, structures previously accessible only through longer, more delicate routes. Comparisons with older cation- and radical-based methods underscored that those approaches either fail outright or lose anomeric control with the most crowded partners, while the new protocol maintains precision.
What this means for future medicines
In everyday terms, this work delivers a reliable “snap connector” for joining complex sugars to bulky drug fragments in a chosen three-dimensional orientation, even at extremely crowded sites. By revealing that the active palladium species are halide-bridged dimers and showing how subtle tweaks to the ligand steer the reaction away from wasteful side steps, the study also offers general design rules for future catalysts. Together, these advances expand the toolbox for chemists seeking more stable, selective sugar-modified drugs and should accelerate the discovery of next-generation antibiotics, anticancer agents, and treatments for metabolic disease.
Citation: Yang, B., Chen, S., Han, Y. et al. Ligand-enabled next-generation glycosyl Stille cross-coupling for the stereospecific synthesis of sterically hindered aryl C-glycosides. Nat Commun 17, 3015 (2026). https://doi.org/10.1038/s41467-026-69859-2
Keywords: aryl C-glycosides, glycosyl cross-coupling, palladium catalysis, ligand design, glycomimetic drugs