Clear Sky Science · it
Accoppiamento incrociato Glycosyl-Stille di nuova generazione abilitato da ligandi per la sintesi stereospecifica di aril C-glicosidi stericamente ingombrati
Perché è difficile costruire farmaci legati agli zuccheri
Molti farmaci moderni imitano gli zuccheri complessi che decorano la superficie delle nostre cellule. Quando un frammento planare a forma di anello di un farmaco viene unito direttamente a uno zucchero tramite un robusto legame carbonio–carbonio, il risultato—un aril C-glicoside—può essere molto più stabile nell’organismo rispetto a un normale legame zuccherino. Queste strutture compaiono in antibiotici, agenti antitumorali e nuovi farmaci per il diabete, ma sono notoriamente difficili da preparare, specialmente quando l’anello attaccato è voluminoso e affollato. Questo lavoro descrive un nuovo modo per costruire in modo pulito e affidabile tali connessioni zucchero–anello “stericamente ingombrate”, aprendo la strada a una progettazione più rapida di farmaci a base di zuccheri.
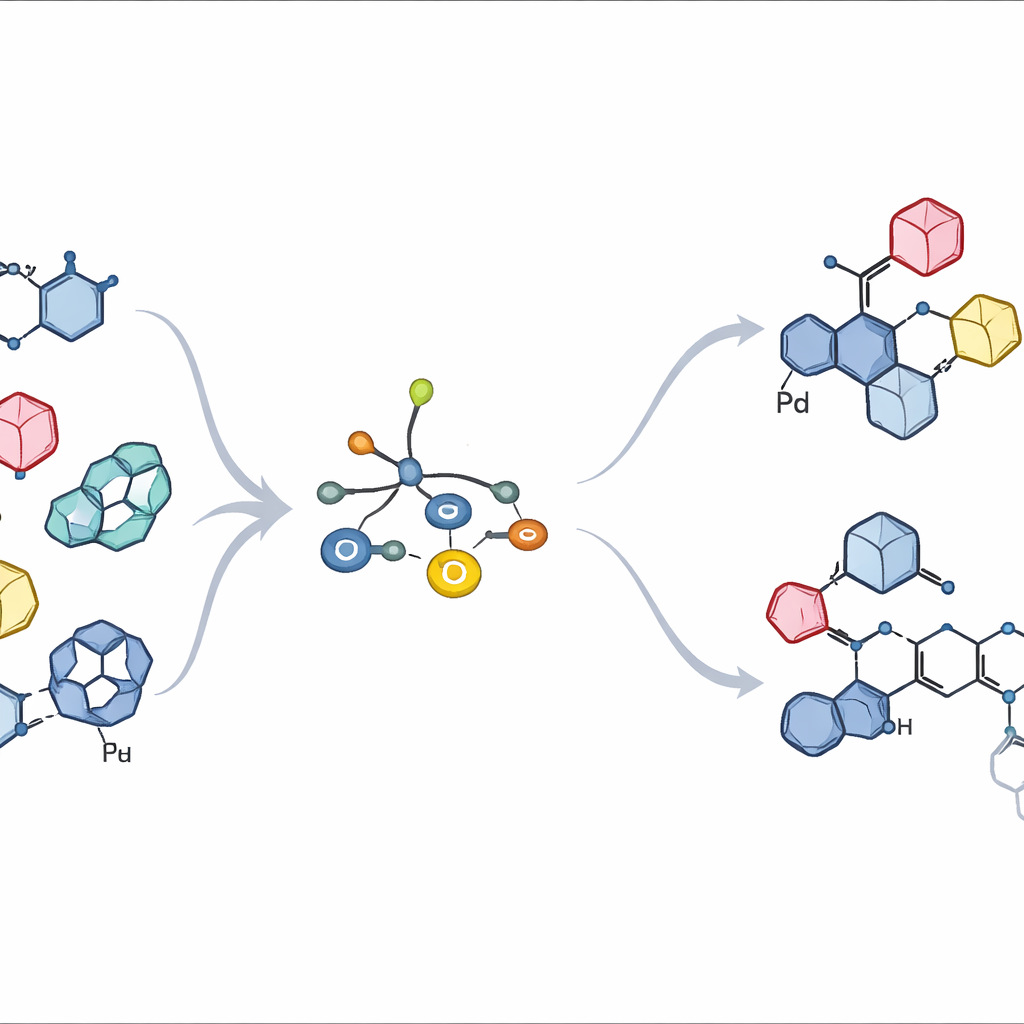
Legami zuccherini ostinati in un giunto affollato
Negli zuccheri naturali, il punto di collegamento chiave è un singolo carbonio alla punta dell’anello, noto come centro anomerico. I chimici devono controllare non solo quale partner si lega lì, ma anche se il nuovo legame punta “verso l’alto” o “verso il basso” nello spazio—due forme che possono comportarsi come molecole completamente diverse in ambito biologico. I metodi tradizionali spesso passano attraverso frammenti zuccherini altamente reattivi e carichi positivamente o tramite intermedi radicalici. Queste vie possono favorire una sola direzione, alterare la geometria di partenza o generare una miscela di prodotti secondari indesiderati, soprattutto quando l’anello partner è grande, rigido e sostituito con altri gruppi. Di conseguenza, molti aril C-glicosidi preziosi restano fuori portata o richiedono sintesi lunghe e poco efficienti.
Progettare un aiutante metallico più intelligente
Gli autori si basano su un’idea più recente: usare uno zucchero portatore di stagno che ceda il suo carbonio direttamente a un catalizzatore di palladio in un cosiddetto accoppiamento incrociato di Stille. In lavori precedenti, una famiglia di ligandi fosfinici attorno al palladio permetteva a zuccheri contenenti stagno di reagire con anelli aromatici semplici preservando perfettamente l’orientazione originale verso l’alto o verso il basso. Tuttavia, quando il partner aromatico diventava voluminoso—come quelli presenti nei veri farmaci—lo stesso sistema rallentava o si bloccava. Qui, il gruppo ha riprogettato sistematicamente lo scheletro del ligando, sintonizzandone sia l’ingombro sia la capacità di attrarre elettroni. Due nuovi ligandi, denominati L10 e L11, sono emersi come chiari vincitori. Presentano gruppi arilici fluorurati fortemente elettron-attrattori legati al fosforo e braccia contenenti ossigeno posizionate con cura sugli anelli arilici di supporto, che insieme rendono il palladio più incline ad accettare lo zucchero pur riuscendo a rilasciare il prodotto finale.
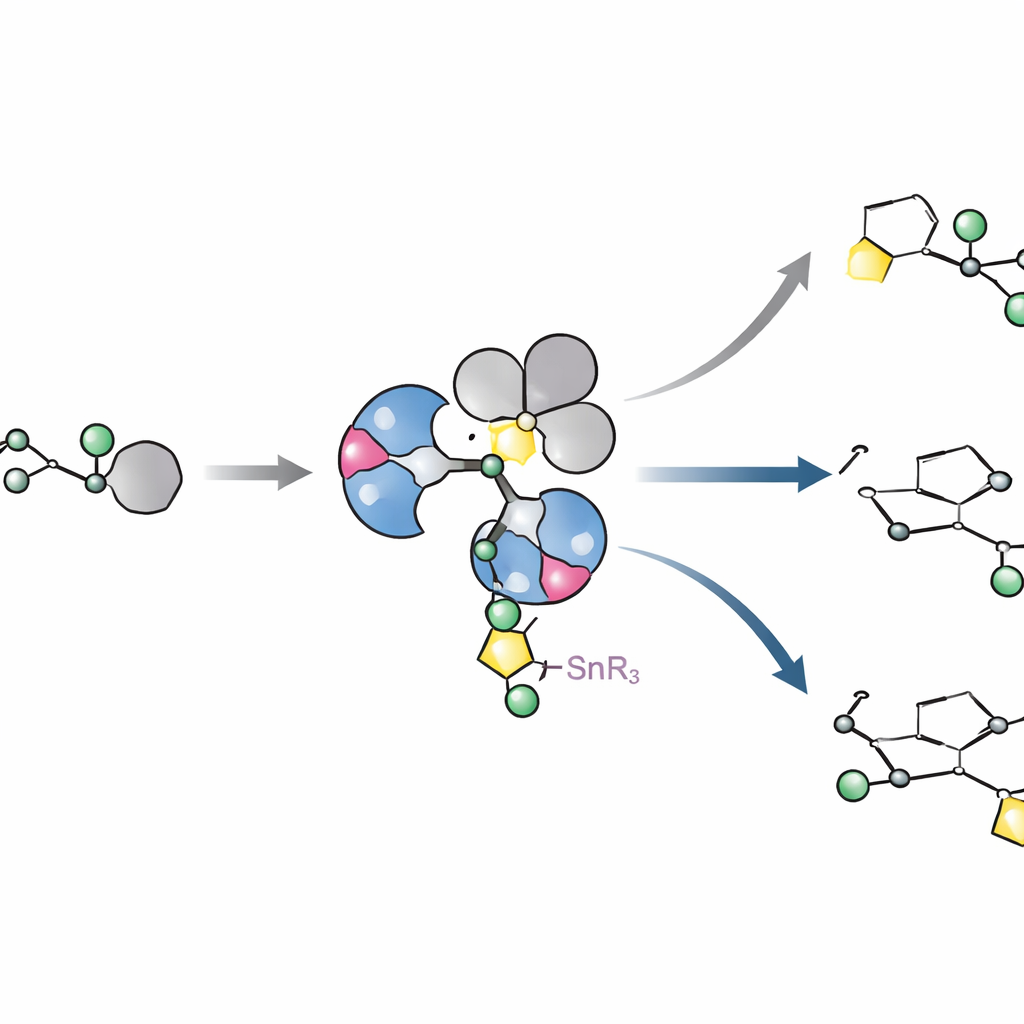
Vedere come appare e si comporta davvero il catalizzatore
Per capire perché questi ligandi funzionano così bene, gli autori hanno isolato e cristallizzato complessi chiave di palladio formati con alogenuro arilico ingombrato. Con sorpresa, invece delle semplici strutture monometalliche a lungo ipotizzate, hanno osservato costantemente dimeri: due centri di palladio bridgiati da ioni alogenuro. Questo arrangiamento si è rivelato una caratteristica naturale dell’intera classe di ligandi, non un’eccezione rara. Esperimenti cinetici e calcoli quantochimici hanno quindi tracciato come questo dimero si apra quanto basta per reagire con lo zucchero portatore di stagno, trasferisca il frammento zuccherino in un singolo passaggio che preserva la geometria e infine formi il legame carbonio–carbonio desiderato. I calcoli hanno mostrato che L11 sposta sottilmente la densità elettronica in modo che la formazione del legame sia favorita rispetto a una via concorrente che troncherebbe un gruppo metossile e produrrebbe spreco di materiale.
Da reazioni modello a obiettivi reali simili a farmaci
Forte di queste intuizioni, il gruppo ha testato una vasta gamma di partner aromatici affollati e donatori zuccherini. Più di 65 esempi—inclusi zuccheri protetti, deossi-zuccheri e persino brevi catene zuccherine—sono stati uniti con rese da buone a eccellenti con controllo praticamente perfetto sull’orientazione anomerica finale. Notevolmente, anche zuccheri completamente non protetti, che portano molteplici gruppi ossidrile liberi, hanno potuto essere accoppiati in modo pulito, cosa che di solito causa gravi reazioni collaterali. Il metodo ha funzionato anche nella modifica in fase avanzata di farmaci approvati e molecole bioattive: installando prima un singolo atomo di alogeno sull’anello aromatico di un farmaco e poi applicando il nuovo accoppiamento, gli autori hanno agganciato unità zuccherine per creare nuovi analoghi in una o due sole tappe.
Nuove scorciatoie verso ibridi complessi zucchero–farmaco
Per dimostrare la potenza dell’approccio, gli autori hanno snellito la sintesi del farmaco per il diabete Enavogliflozin e di varianti correlate, formando direttamente il suo ingombrato legame zucchero–arilico in un’unica tappa e ottenendo entrambi i possibili anomeri. Hanno anche preparato sistemi ad anello fuso in cui lo zucchero e l’unità aromatica condividono un nuovo anello, strutture precedentemente accessibili solo tramite vie più lunghe e delicate. I confronti con metodi più datati basati su cationi e radicali hanno evidenziato che quelle strategie o falliscono del tutto o perdono il controllo anomerico con i partner più affollati, mentre il nuovo protocollo mantiene la precisione.
Cosa significa per i farmaci futuri
In termini pratici, questo lavoro fornisce un «connettore a scatto» affidabile per unire zuccheri complessi a frammenti di farmaci voluminosi in una data orientazione tridimensionale, anche in siti estremamente affollati. Rivelando che le specie attive di palladio sono dimeri bridgiati da alogenuro e mostrando come sottili modifiche al ligando dirigano la reazione lontano da vie collaterali sprecone, lo studio offre anche regole generali di progettazione per futuri catalizzatori. Insieme, questi progressi ampliano la cassetta degli attrezzi per i chimici che cercano farmaci modificati con zuccheri più stabili e selettivi e dovrebbero accelerare la scoperta di antibiotici di nuova generazione, agenti antitumorali e terapie per le malattie metaboliche.
Citazione: Yang, B., Chen, S., Han, Y. et al. Ligand-enabled next-generation glycosyl Stille cross-coupling for the stereospecific synthesis of sterically hindered aryl C-glycosides. Nat Commun 17, 3015 (2026). https://doi.org/10.1038/s41467-026-69859-2
Parole chiave: aril C-glicosidi, accoppiamento incrociato glicosilico, catalisi al palladio, progettazione del ligando, farmaci glicomimetici