Clear Sky Science · de
Ligand-gestützte Next-Generation-Glycosyl‑Stille-Kupplung für die stereospezifische Synthese sterisch gehinderter arylischer C‑Glykoside
Warum es schwer ist, zuckerverknüpfte Medikamente zu bauen
Viele moderne Medikamente ahmen die komplexen Zucker nach, die die Oberfläche unserer Zellen schmücken. Wenn ein flacher, ringförmiger Fragment eines Wirkstoffs direkt über eine stabile Kohlenstoff–Kohlenstoff‑Bindung an einen Zucker gekoppelt wird, entsteht ein arylisches C‑Glykosid, das im Körper weitaus beständiger sein kann als eine normale Zuckerverknüpfung. Solche Strukturen finden sich in Antibiotika, Krebsmedikamenten und neuen Diabetesmitteln, sind aber notorisch schwer herzustellen — insbesondere wenn der angehängte Ring sperrig und dicht substituiert ist. Dieses Papier beschreibt eine neue Methode, um solche „sterisch gehinderten“ Zucker‑Ring‑Verknüpfungen sauber und zuverlässig zu erzeugen und damit die Entwicklung zuckerbasierter Medikamente zu beschleunigen.
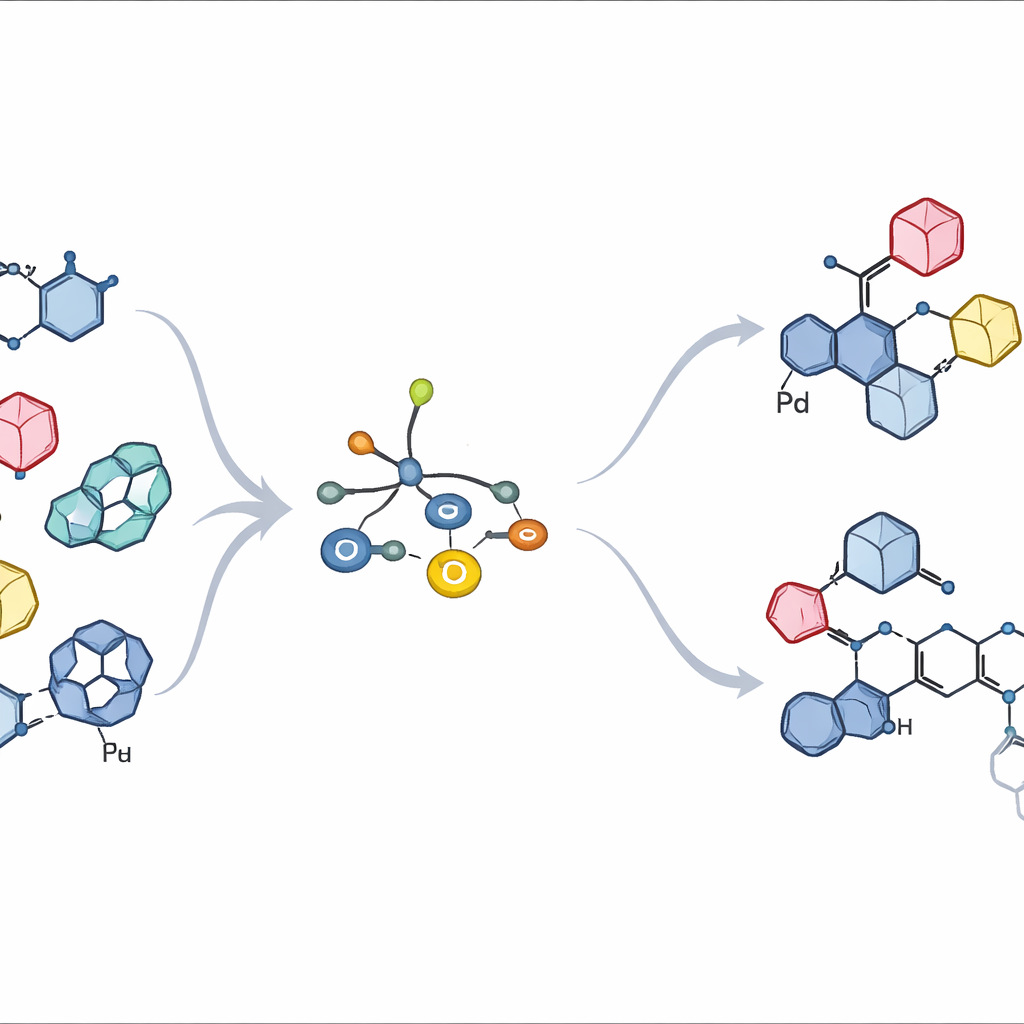
Hartnäckige Zuckerbindungen an einem überfüllten Bindungspunkt
Bei natürlichen Zuckern liegt der entscheidende Bindungspunkt an einem einzelnen Kohlenstoff an der Spitze des Rings, dem sogenannten anomeren Zentrum. Chemiker müssen nicht nur kontrollieren, welcher Partner dort bindet, sondern auch, ob die neue Bindung im Raum „nach oben“ oder „nach unten“ zeigt — zwei Formen, die sich biologisch ganz unterschiedlich verhalten können. Traditionelle Methoden verlaufen oft über hochreaktive, positiv geladene Zuckerfragmente oder radikalische Zwischenstufen. Diese Wege können nur eine Richtung begünstigen, die Ausgangsgeometrie verwischen oder eine Mischung unerwünschter Nebenprodukte erzeugen, besonders wenn der aromatische Partner groß, starr und mit weiteren Gruppen versehen ist. Daher bleiben viele wertvolle arylische C‑Glykoside unerreichbar oder erfordern lange, verschwenderische Synthesen.
Entwurf eines schlaueren Metallhelfers
Die Autoren bauen auf einer neueren Idee auf: Einen zinnhaltigen Zucker zu verwenden, der sein Kohlenstoffatom direkt an einen Palladiumkatalysator in einer sogenannten Stille‑Kreuzkupplung übergibt. In früheren Arbeiten erlaubte eine Familie von Phosphinliganden um Palladium herum, dass Zinn‑Zucker mit einfachen aromatischen Ringen gekoppelt werden konnten und dabei die ursprüngliche oben‑/unten‑Orientierung perfekt beibehielten. Wenn der aromatische Partner jedoch sperrig wurde — wie in echten Wirkstoffen — verlangsamte oder stoppte dasselbe System. Hier hat das Team das Ligandengerüst systematisch neu gestaltet und sowohl dessen Raumanspruch als auch dessen elektronenziehende Eigenschaften feinjustiert. Zwei neue Liganden, L10 und L11 genannt, erwiesen sich als klare Sieger. Sie besitzen stark elektronenziehende, fluorierte Arylgruppen am Phosphor und sorgfältig platzierte, sauerstoffhaltige Arme an den stützenden Arylringen, die zusammen Palladium anfälliger machen, den Zucker aufzunehmen, gleichzeitig aber die Freisetzung des Endprodukts ermöglichen.
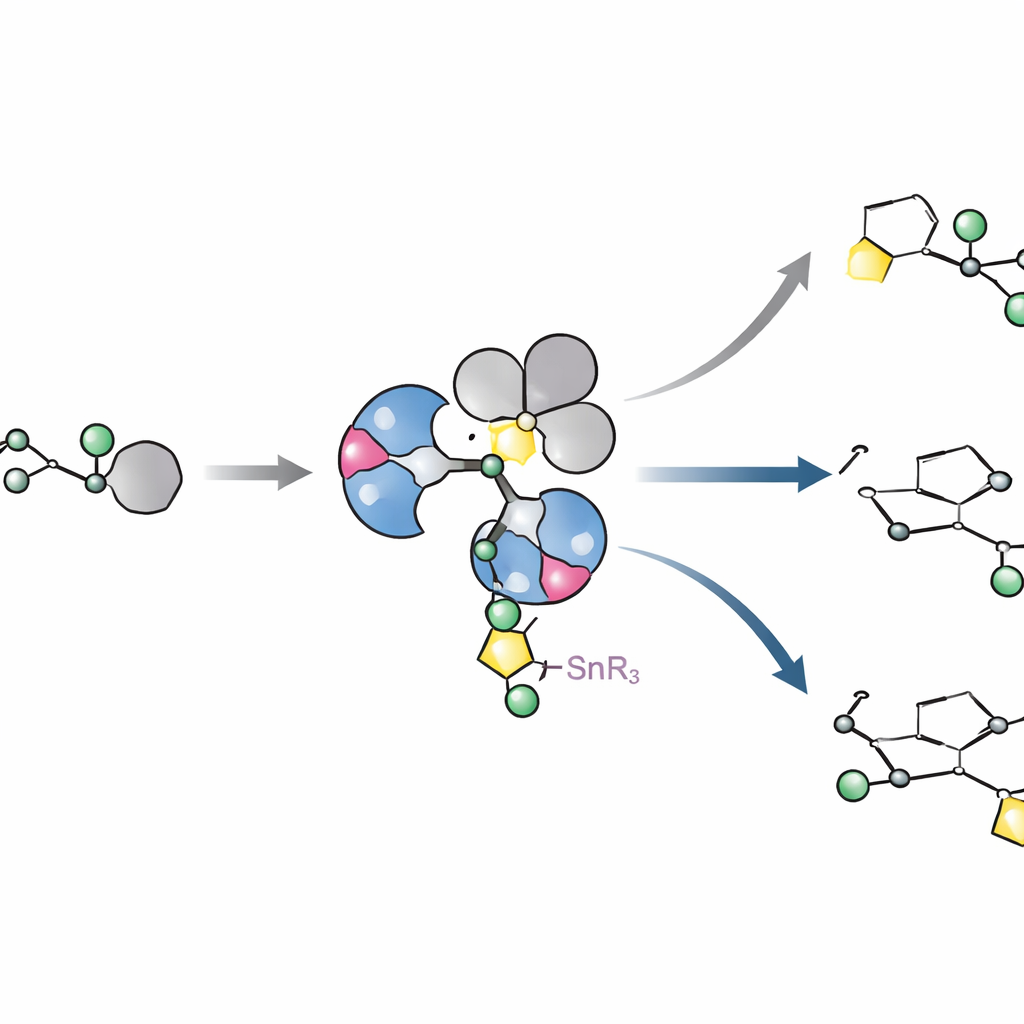
Wie der Katalysator wirklich aussieht und sich verhält
Um zu verstehen, warum diese Liganden so gut funktionieren, isolierten und kristallisierten die Autoren Schlüsselkomplexe von Palladium, die mit sperrigen arylischen Halogeniden gebildet werden. Überraschenderweise beobachteten sie statt der einfachen monomeren Metallstrukturen, die lange angenommen worden waren, konsistent Dimere: zwei Palladiumzentren, die durch Halogenidionen überbrückt sind. Diese Anordnung erwies sich als charakteristisches Merkmal der gesamten Ligandklasse, nicht als seltene Ausnahme. Kinetische Experimente und quantenchemische Berechnungen verfolgten dann, wie dieses Dimer gerade so weit auseinanderbricht, dass es mit dem zinnhaltigen Zucker reagiert, das Zuckerfragment in einem einzigen, geometrieerhaltenden Schritt überträgt und schließlich die gewünschte Kohlenstoff–Kohlenstoff‑Bindung bildet. Die Rechnungen zeigten, dass L11 die Elektronendichte so fein verschiebt, dass die Bindungsbildung gegenüber einem konkurrierenden Weg bevorzugt wird, der eine Methoxygruppe abschneiden und Material verschwenden würde.
Von Modellreaktionen zu echten wirkstoffähnlichen Zielen
Gestützt auf diese Erkenntnisse testete das Team eine breite Palette sperriger aromatischer Partner und Zuckerspender. Mehr als 65 Beispiele — einschließlich geschützter Zucker, deoxy‑Zucker und sogar kurzer Zuckerketten — wurden in guten bis exzellenten Ausbeuten gekoppelt, mit im Wesentlichen perfekter Kontrolle darüber, ob die finale Bindung nach oben oder nach unten zeigt. Bemerkenswerterweise konnten sogar vollständig ungeschützte Zucker, die mehrere freie Hydroxylgruppen tragen, sauber gekoppelt werden — etwas, das normalerweise schwere Nebenreaktionen verursacht. Die Methode funktionierte auch bei der „late‑stage“‑Modifizierung zugelassener Medikamente und bioaktiver Moleküle: Indem zuerst ein einzelnes Halogenatom in den aromatischen Ring eines Arzneistoffs eingeführt und dann die neue Kupplung angewendet wurde, setzten die Autoren Zuckereinheiten in nur ein oder zwei Schritten auf und erzeugten so neue Analoga.
Neue Abkürzungen zu komplexen Zucker‑Wirkstoff‑Hybriden
Um die Leistungsfähigkeit des Ansatzes zu demonstrieren, strafften die Autoren die Synthese des Diabetesmedikaments Enavogliflozin und verwandter Varianten, indem sie dessen stark überfüllte Zucker‑aryl‑Bindung in einem Schritt direkt schmiedeten und beide möglichen anomeren Formen zugänglich machten. Sie bereiteten auch gefügte Ringsysteme vor, in denen Zucker und aromatische Einheit einen neuen Ring teilen — Strukturen, die zuvor nur über längere, empfindlichere Routen erreichbar waren. Vergleiche mit älteren kationen‑ und radikalbasierten Methoden zeigten, dass diese Ansätze entweder ganz versagen oder bei den am stärksten überfüllten Partnern die anomere Kontrolle verlieren, während das neue Protokoll Präzision bewahrt.
Was das für zukünftige Medikamente bedeutet
Alltäglich ausgedrückt liefert diese Arbeit einen zuverlässigen „Schnappverbinder“, um komplexe Zucker mit sperrigen Wirkstofffragmenten in einer gewählten dreidimensionalen Orientierung zu verbinden, selbst an extrem überfüllten Stellen. Indem sie zeigen, dass die aktiven Palladiumspezies halogenid‑brückende Dimere sind und wie subtile Anpassungen am Liganden die Reaktion von verschwenderischen Nebenwegen weglenken, bieten die Autoren außerdem allgemeine Designregeln für zukünftige Katalysatoren. Zusammen erweitern diese Fortschritte das Werkzeugset für Chemiker, die stabilere, selektivere zuckermodifizierte Wirkstoffe anstreben, und sollten die Entdeckung von Next‑Generation‑Antibiotika, Krebsmedikamenten und Therapien gegen Stoffwechselerkrankungen beschleunigen.
Zitation: Yang, B., Chen, S., Han, Y. et al. Ligand-enabled next-generation glycosyl Stille cross-coupling for the stereospecific synthesis of sterically hindered aryl C-glycosides. Nat Commun 17, 3015 (2026). https://doi.org/10.1038/s41467-026-69859-2
Schlüsselwörter: arylische C‑Glykoside, glycosylische Kreuzkupplung, Palladiumkatalyse, Ligandendesign, Glykomimetische Medikamente