Clear Sky Science · pl
Zwiększanie wrażliwości na inhibitory kinazy tyrozynowej poprzez przywrócenie aktywności IKAROS na ekspresję GLUT1 i glikolizę w ostrym limfoblastycznym białaczce z chromosomem Filadelfia
Dlaczego to badanie nad rakiem krwi ma znaczenie
Niektórzy pacjenci z poważnym nowotworem krwi, zwanym ostrą białaczką limfoblastyczną z chromosomem Filadelfia (Ph+ ALL), przestają dobrze reagować na nowoczesne leki ukierunkowane, nawet na najsilniejsze z nich. Badanie to wyjaśnia, dlaczego podzbiór tych białaczek przeżywa mimo leczenia, i pokazuje potencjalny sposób przywrócenia wrażliwości za pomocą drugiego leku, który uderza w linię zaopatrzenia nowotworu w energię. Praca wskazuje na terapie skojarzone, które mogłyby pomóc pacjentom, u których dochodzi do nawrotu po wyczerpaniu standardowych opcji.
Oporny rodzaj białaczki
Ph+ ALL jest napędzana przez nieprawidłowe białko fuzyjne, które działa jak przyspieszony pedał gazu, zmuszając młode białe krwinki do niekontrolowanego wzrostu. Leki zwane inhibitorami kinaz tyrozynowych (TKI), takie jak imatynib i ponatynib, zostały zaprojektowane, by trafić w ten wadliwy „akcelerator” i znacznie poprawiły przeżywalność. Mimo to wielu dorosłych nadal doświadcza nawrotów. Cechą wspólną tych trudnych przypadków jest często uszkodzenie genu o nazwie IKZF1, który koduje białko IKAROS, normalnie pomagające kontrolować komórki limfoidalne. Gdy IKAROS jest utracony lub nieaktywny, pacjenci mają gorsze rokowania i często nie odpowiadają trwale na TKI, terapie immunologiczne ani przeszczepy.
Jak komórki białaczki omijają zasady energetyczne
Komórki nowotworowe często przepinają sposób pozyskiwania energii, faworyzując szybką, acz mało wydajną ścieżkę zwaną glikolizą, nawet gdy tlenu jest pod dostatkiem. Autorzy wykazali, że komórki Ph+ ALL z częstym delecją IKZF1 (nieprawidłowa wersja nazwana Ik6) były mniej wrażliwe na TKI niż komórki z nienaruszonym IKZF1. Te oporne komórki wykazywały wysokie poziomy GLUT1, białka transportującego glukozę do wnętrza komórki, oraz zwiększoną glikolizę. Gdy badacze zmniejszyli ilość GLUT1 w komórkach białaczki, zużycie glukozy i produkcja kwasu mlekowego spadły, wzrost komórek zwolnił, a więcej komórek weszło na ścieżkę zaprogramowanej śmierci. W próbkach od pacjentów GLUT1 był wyraźnie wyższy w szpiku kostnym chorych z Ph+ ALL niż u zdrowych dawców, a pacjenci z najwyższymi poziomami GLUT1 częściej doświadczali nawrotów i mieli krótsze przeżycie.
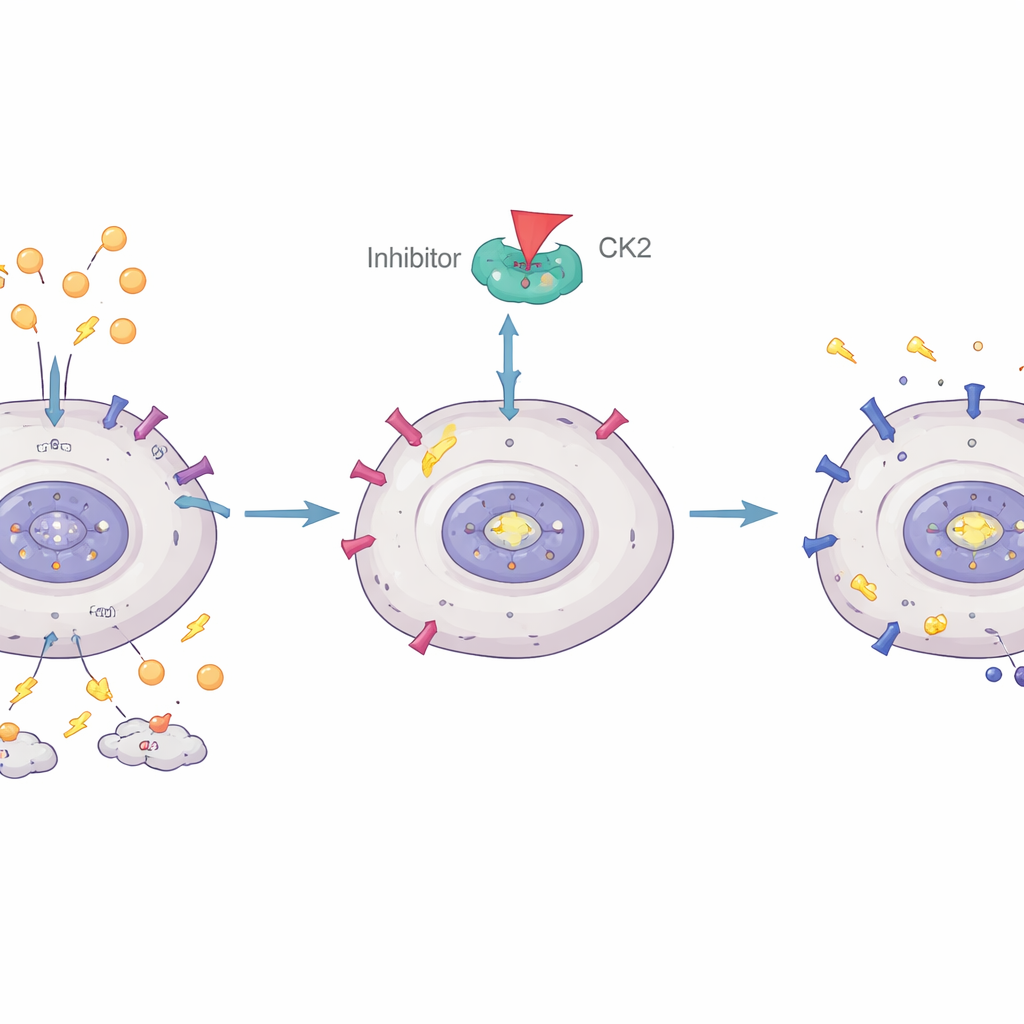
Przywracanie utrzymanego hamulca w komórkach białaczki
Zespół zapytał następnie, czy da się przywrócić supresyjną rolę IKAROS. We wcześniejszych pracach pokazali, że enzym zwany kinazą CK2 (casein kinase II) może chemicznie modyfikować IKAROS w sposób osłabiający jego zdolność do kontroli genów. Tutaj zastosowali lek blokujący CK2 o nazwie CX-4945. W hodowlach komórek Ph+ ALL zwiększenie poziomów IKAROS spowalniało podziały komórek i zwiększało śmierć komórkową. Sam CX-4945 również hamował wzrost, ale był szczególnie silny w połączeniu z nadekspresją IKAROS, co wskazuje, że blokada CK2 pomaga IKAROS pełnić rolę hamulca w białaczce.
Terapia skojarzona celująca w wzrost i paliwo
Następnie badacze leczyli komórki białaczki i pierwotne komórki od pacjentów kombinacją TKI i CX-4945. Parom leków (imatynib lub ponatynib plus CX-4945) udało się zatrzymać wzrost komórek i wywołać ich śmierć skuteczniej niż któremukolwiek z leków z osobna; formalne analizy synergii potwierdziły, że efekt kombinacji był większy niż suma efektów poszczególnych składników. Co ważne, zarówno w standardowym modelu białaczki u myszy, jak i w bardziej realistycznym modelu xenotransplantacyjnym pochodzącym od pacjenta (z wyjątkowo trudnym do leczenia Ik6+ Ph+ ALL), te kombinacje zmniejszały masę nowotworu, redukowały powiększenie śledziony i znacząco wydłużały przeżycie. Na poziomie molekularnym kombinacje obniżały poziomy GLUT1, zmniejszały zużycie glukozy i wydzielanie kwasu mlekowego oraz tłumiły mierniki glikolizy w czasie rzeczywistym. Genetyczne stłumienie GLUT1 dodatkowo spotęgowało efekt kombinacji leków, co podkreśla, że zablokowanie tej drogi paliwowej jest kluczowe dla ich działania.
Powiązanie IKAROS z "bramką" cukru nowotworu
Pogłębiając analizę, autorzy pokazali, że IKAROS bezpośrednio wiąże się z regionem kontrolnym genu GLUT1 i go wycisza. Gdy CK2 był hamowany przez CX-4945, przyczepność IKAROS do promotora GLUT1 wzmacniała się, poziomy GLUT1 spadały, a glikoliza malała. Sama nadekspresja IKAROS była wystarczająca, by zmniejszyć użycie glukozy i produkcję kwasu mlekowego, a CX-4945 wzmacniał ten efekt. Wyniki te stawiają IKAROS jako bezpośredniego regulatora ilości glukozy, jaką komórki Ph+ ALL mogą importować, łącząc istotne uszkodzenie genetyczne ze zmienionym metabolizmem nowotworu i opornością na leki.
Co to może znaczyć dla pacjentów
Mówiąc prościej, badanie sugeruje, że niektóre komórki Ph+ ALL przetrwają terapię TKI zarówno przez utratę genetycznego hamulca (IKAROS), jak i przez zwiększenie „bramki” dla cukru (GLUT1), która zasila ich zmieniony metabolizm. Poprzez blokadę CK2 za pomocą CX-4945 badacze byli w stanie przywrócić aktywność IKAROS, wyłączyć GLUT1, pozbawić komórki białaczki paliwa i sprawić, że TKI działały lepiej w komórkach i modelach mysich, nawet gdy wcześniejsze terapie zawiodły u pierwotnego pacjenta. Choć konieczne będą badania kliniczne, praca wskazuje na przyszłość, w której łączenie TKI z lekami resetującymi wewnętrzne hamulce komórkowe i metabolizm może dać nową nadzieję osobom z oporną Ph+ ALL.
Cytowanie: Zhang, L., Han, Q., Xiang, H. et al. Enhancing tyrosine kinase inhibitor sensitivity by restoring IKAROS activity on GLUT1 expression and glycolysis in Philadelphia chromosome-positive acute lymphoblastic leukemia. Leukemia 40, 794–805 (2026). https://doi.org/10.1038/s41375-026-02898-2
Słowa kluczowe: Ostra białaczka limfoblastyczna z chromosomem Filadelfia, oporność na inhibitory kinazy tyrozynowej, IKZF1 IKAROS, GLUT1 glikoliza, inhibitor CK2 CX-4945