Clear Sky Science · en
Enhancing tyrosine kinase inhibitor sensitivity by restoring IKAROS activity on GLUT1 expression and glycolysis in Philadelphia chromosome-positive acute lymphoblastic leukemia
Why this blood cancer study matters
Some patients with a serious blood cancer called Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL) no longer respond well to modern targeted drugs, even the most powerful ones. This study uncovers why a subset of these leukemias stay alive despite treatment and shows a potential way to re-sensitize them using a second medicine that targets the cancer’s energy supply line. The work points toward combination therapies that could help patients who currently relapse after exhausting standard options.
A stubborn form of leukemia
Ph+ ALL is driven by an abnormal fusion protein that acts like a stuck accelerator, pushing young white blood cells to grow uncontrollably. Drugs called tyrosine kinase inhibitors (TKIs), such as imatinib and ponatinib, are designed to hit this faulty accelerator and have dramatically improved survival. Yet many adults still relapse. A common feature of these difficult cases is damage to a gene called IKZF1, which encodes a protein named IKAROS that normally helps keep lymphoid cells in check. When IKAROS is missing or disabled, patients tend to do worse and often do not respond durably to TKIs, immune therapies, or transplants.
How leukemia cells bend the energy rules
Cancer cells often rewire how they make energy, favoring a fast but wasteful pathway called glycolysis even when oxygen is plentiful. The authors found that Ph+ ALL cells with a frequent IKZF1 deletion (an abnormal version called Ik6) were less sensitive to TKIs than cells with intact IKZF1. These resistant cells showed high levels of GLUT1, a protein that ferries glucose into the cell, and they displayed heightened glycolysis. When the researchers reduced GLUT1 in leukemia cells, glucose use and lactic acid production fell, cell growth slowed, and more cells underwent programmed death. In patient samples, GLUT1 was markedly higher in Ph+ ALL bone marrow than in healthy donors, and patients with the highest GLUT1 levels relapsed more often and had shorter survival.
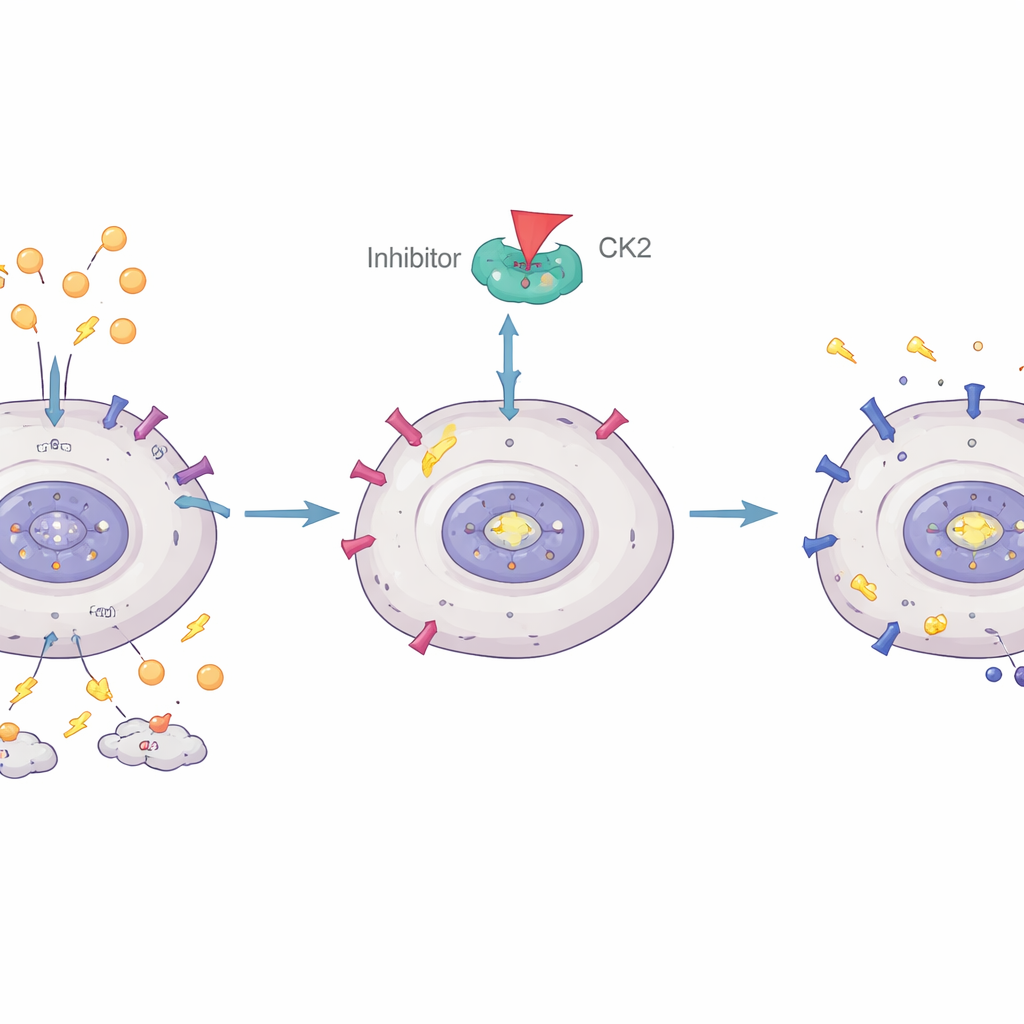
Restoring a lost brake in leukemia cells
The team then asked whether they could restore IKAROS’s tumor-suppressing activity. In earlier work, they had shown that an enzyme called casein kinase II (CK2) can chemically modify IKAROS in a way that weakens its ability to control genes. Here, they used a CK2-blocking drug called CX-4945. In cultured Ph+ ALL cells, boosting IKAROS levels slowed cell division and increased cell death. CX-4945 on its own also curbed growth, but it was especially powerful when combined with IKAROS overexpression, indicating that blocking CK2 helps IKAROS function as a brake on leukemia.
Combination therapy that targets growth and fuel
Next, the researchers treated leukemia cells and primary patient cells with TKIs together with CX-4945. The drug pairs (imatinib or ponatinib plus CX-4945) stopped cell growth and triggered cell death more effectively than either drug alone, with formal synergy analyses confirming that the whole was greater than the sum of its parts. Importantly, in both a standard mouse leukemia model and a more realistic patient-derived xenograft built from an especially hard-to-treat Ik6+ Ph+ ALL sample, these combinations shrank leukemia burdens, reduced spleen enlargement, and significantly extended survival. On a molecular level, the combinations lowered GLUT1 levels, reduced glucose consumption and lactic acid output, and dampened real-time measures of glycolysis. Genetic knockdown of GLUT1 further heightened the impact of the drug combinations, underscoring that shutting down this fuel route is central to their effect.
Linking IKAROS to the cancer’s sugar gate
Delving deeper, the investigators showed that IKAROS directly binds to the control region of the GLUT1 gene and turns it down. When CK2 was inhibited with CX-4945, IKAROS’s grip on the GLUT1 promoter strengthened, GLUT1 levels dropped, and glycolysis declined. Overexpressing IKAROS alone was enough to reduce glucose use and lactic acid production, and CX-4945 amplified this effect. These findings position IKAROS as a direct regulator of how much glucose Ph+ ALL cells can import, connecting a key genetic lesion to the cancer’s altered metabolism and drug resistance.
What this could mean for patients
In simple terms, this study suggests that certain Ph+ ALL cells survive TKIs by both losing a genetic brake (IKAROS) and turning up a sugar gate (GLUT1) that feeds their altered metabolism. By blocking CK2 with CX-4945, the researchers were able to restore IKAROS activity, shut down GLUT1, starve leukemia cells of fuel, and make TKIs work better in cells and mouse models, even when prior therapies had failed the original patient. While clinical trials will be needed, the work points to a future in which pairing TKIs with drugs that reset the cell’s internal brakes and energy use could offer new hope for people with resistant Ph+ ALL.
Citation: Zhang, L., Han, Q., Xiang, H. et al. Enhancing tyrosine kinase inhibitor sensitivity by restoring IKAROS activity on GLUT1 expression and glycolysis in Philadelphia chromosome-positive acute lymphoblastic leukemia. Leukemia 40, 794–805 (2026). https://doi.org/10.1038/s41375-026-02898-2
Keywords: Philadelphia chromosome-positive ALL, tyrosine kinase inhibitor resistance, IKZF1 IKAROS, GLUT1 glycolysis, CK2 inhibitor CX-4945