Clear Sky Science · nl
Een nieuwe diep-intronische COL5A1‑variant in een familie met het Ehlers‑Danlos‑syndroom: functionele karakterisering met een minigene‑assay
Verborgen aanwijzingen in rekbare huid
Waarom krijgen sommige mensen gemakkelijk blauwe plekken, littekens die diep insnijden en ongebruikelijk rekbare huid en soepele gewrichten? Voor families die leven met het Ehlers‑Danlos‑syndroom kunnen deze alledaagse problemen pijnlijk en verwarrend zijn. Deze studie volgt zo’n familie in China en laat zien hoe één goed verborgen verandering in hun DNA het interne geraamte van het lichaam kan verzwakken, en zo nieuwe inzichten biedt voor diagnose en toekomstige behandelideeën voor deze zeldzame aandoening.
Wat deze familie doormaakte
Het onderzoek concentreert zich op een 30‑jarige vrouw die vanwege langdurige huid‑ en gewrichtsklachten een dermatologische polikliniek bezocht. Ze had zachte, fluweelachtige huid die meer dan gebruikelijk kon uitrekken, genas met dunne, papierachtige littekens en kreeg gemakkelijk blauwe plekken. Haar vingergewrichten kon ze verder terug buigen dan normaal. Haar vader had losse huid en enkele gelaatstrekken die vaak bij deze aandoening voorkomen, terwijl haar moeder geen symptomen had. Samen wezen hun verschijnselen op het klassieke type van het Ehlers‑Danlos‑syndroom, een vorm die vooral de huid en gewrichten aantast maar ook vragen kan oproepen over bloedvaten en andere organen.
Zoeken naar antwoorden in de genen
Het klassieke Ehlers‑Danlos‑syndroom hangt meestal samen met veranderingen in genen die collageen bouwen, de eiwitketens die huid en veel andere weefsels versterken. Het team gebruikte medische whole‑exome sequencing, die duizenden ziektegerelateerde genen scant, om verdachte veranderingen te zoeken bij de patiënte en haar ouders. Ze vonden een subtiele wijziging in een gen genaamd COL5A1, dat helpt bij de productie van type V collageen. Deze verandering lag niet in het gebruikelijke eiwitcoderende deel van het gen, maar diep in een niet‑coderend segment. Op het eerste gezicht was zo’n verborgen wijziging moeilijk te classificeren, dus moesten de onderzoekers testen of het daadwerkelijk verstoorde hoe het gen gelezen werd.
Een labtest die het genlezen nabootst
Om het effect van deze verborgen DNA‑verandering te onderzoeken, gebruikten de onderzoekers een instrument dat bekendstaat als een minigene‑assay. Ze bouwden twee miniatuurversies van het COL5A1‑gensegment: één met de gebruikelijke sequentie en één met de variant van de familie. Deze constructen werden in menselijke cellen in het laboratorium ingebracht om te zien hoe de cellulaire machinerie ze zou verwerken. De normale versie produceerde het verwachte RNA‑bericht, terwijl de gewijzigde versie twee berichten opleverde. Eén zag er normaal uit, maar in het andere was een extra ingevoegd stuk aanwezig, een zogenoemde pseudoexon, afkomstig uit wat stil had moeten blijven in het genetisch materiaal.
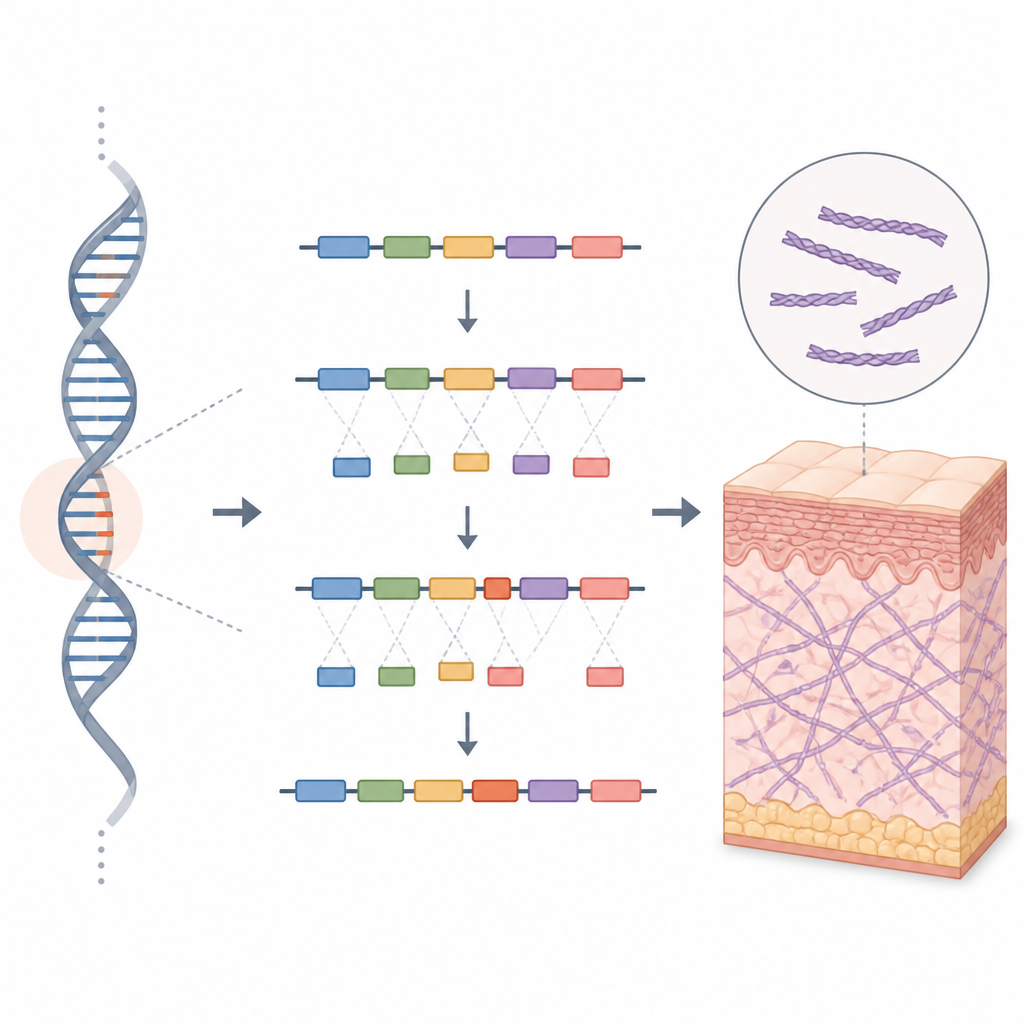
Hoe een klein extra stukje kwetsbaar weefsel veroorzaakt
Dit extra ingevoegde stukje verschuift het leesraam van het genische bericht en introduceert snel een stopsignaal dat het eiwit voortijdig afbreekt. Het resultaat zal naar verwachting óf een ingekort, niet‑werkend fragment van het COL5A1‑eiwit zijn, óf helemaal geen eiwit, omdat cellen dergelijke foutieve boodschappen vaak afbreken. In beide gevallen heeft het lichaam te weinig functioneel type V collageen, waardoor het collageennetwerk dat huid en gewrichten ondersteunt verzwakt. Het familiale patroon, het ontbreken van deze variant in populatiedatabanken en het duidelijke laboratoriumbewijs voor foutieve splicing leidden de auteurs ertoe de verandering als waarschijnlijk ziekteveroorzakend te classificeren volgens de huidige richtlijnen voor klinische genetica.
Waarom deze verborgen wijziging ertoe doet
Dit werk toont aan dat belangrijke ziekteveroorzakende veranderingen zich kunnen verschuilen veraf van de gebruikelijke, eenvoudig te scannen delen van genen, en dat gespecialiseerde RNA‑niveau tests nodig kunnen zijn wanneer routinematige genetische schermen een sterke klinische aanwijzing niet verklaren. Het wijst ook op toekomstige behandelingen die in principe dit soort fouten zouden kunnen corrigeren door het extra pseudoexon te blokkeren en het normale bericht te herstellen, bijvoorbeeld met antisense‑oligonucleotiden. Voor mensen met het klassieke Ehlers‑Danlos‑syndroom verdiept studie als deze het begrip van waarom hun weefsels kwetsbaar zijn en kan het uiteindelijk de weg openen naar preciezere diagnoses en gerichte therapieën.
Bronvermelding: Zhao, J., Feng, J. A novel deep intronic COL5A1 variant in an Ehlers-Danlos syndrome family: functional characterization by minigene assay. Sci Rep 16, 15232 (2026). https://doi.org/10.1038/s41598-026-46346-8
Trefwoorden: Ehlers‑Danlos‑syndroom, COL5A1, intronische variant, aberrante splicing, bindweefsel