Clear Sky Science · de
Eine neuartige tief-intronische COL5A1-Variante in einer Familie mit Ehlers-Danlos-Syndrom: funktionelle Charakterisierung mittels Minigen-Assay
Verborgene Hinweise in dehnbarer Haut
Warum bekommen manche Menschen leicht blaue Flecken, vernarben tief und haben ungewöhnlich dehnbare Haut sowie bewegliche Gelenke? Für Familien, die mit dem Ehlers-Danlos-Syndrom leben, können diese Alltagsprobleme schmerzhaft und verwirrend sein. Diese Studie begleitet eine solche Familie in China und zeigt, wie eine einzige, gut versteckte Veränderung in ihrer DNA das innere Gerüst des Körpers schwächen kann. Sie bietet neue Einblicke in die Diagnostik und mögliche zukünftige Behandlungsansätze für diese seltene Erkrankung.
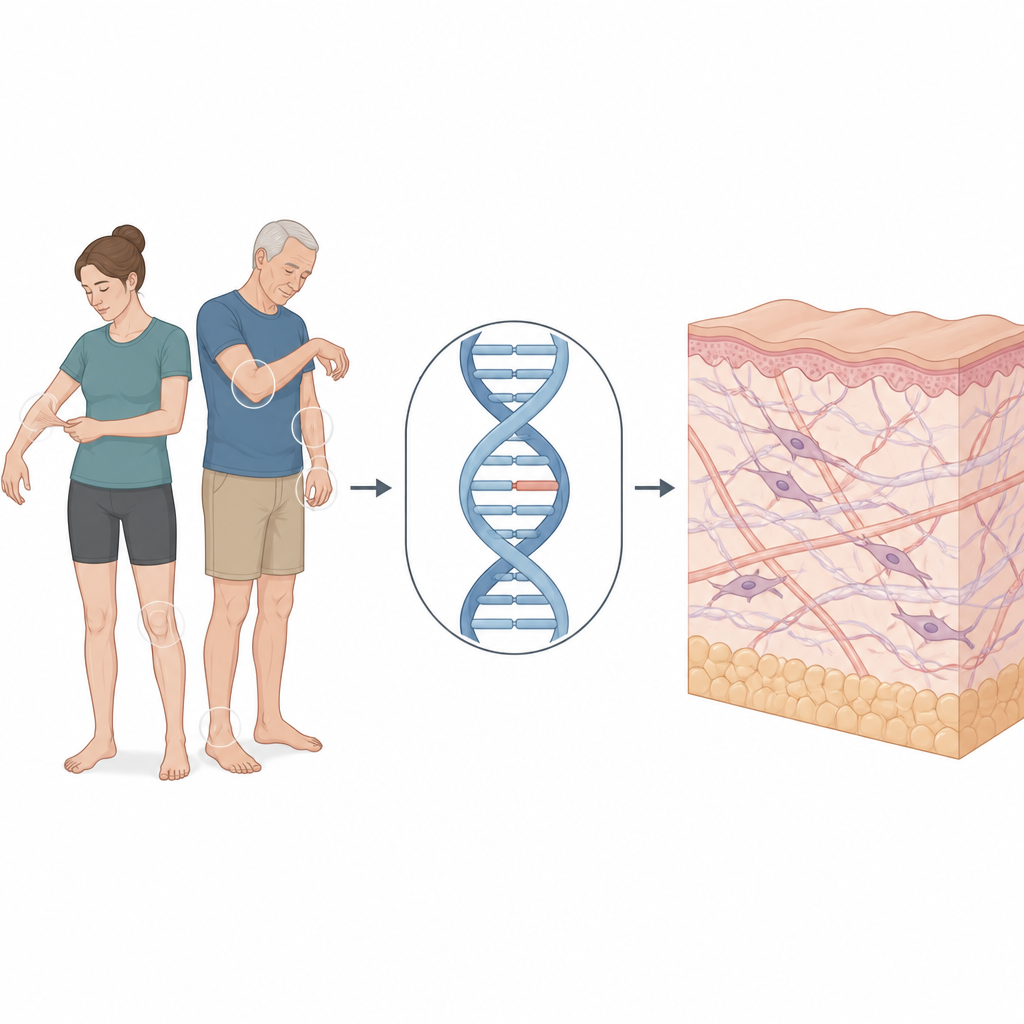
Womit diese Familie konfrontiert war
Der Forschungsfokus liegt auf einer 30-jährigen Frau, die wegen langjähriger Haut- und Gelenkprobleme eine dermatologische Klinik aufsuchte. Sie hatte weiche, samtige Haut, die stärker als üblich dehnbar war, heilte mit dünnen, papierartigen Narben und bekam leicht blaue Flecken. Ihre Fingergelenke ließen sich weiter nach hinten beugen als normal. Ihr Vater zeigte lockere Haut und einige Gesichtszüge, die bei dieser Erkrankung häufig vorkommen, während ihre Mutter keine Symptome hatte. Zusammen deuteten ihre Befunde auf den klassischen Typ des Ehlers-Danlos-Syndroms hin — eine Form, die hauptsächlich Haut und Gelenke betrifft, aber auch Bedenken hinsichtlich Blutgefäßen und anderer Organe aufwerfen kann.
Auf der Suche nach genetischen Antworten
Das klassische Ehlers-Danlos-Syndrom ist meist mit Veränderungen in Genen verbunden, die Kollagen bilden — die Proteinstränge, die Haut und viele andere Gewebe stärken. Das Team nutzte medizinisches Whole-Exome-Sequencing, das Tausende krankheitsrelevante Gene durchsucht, um bei der Patientin und ihren Eltern nach verdächtigen Veränderungen zu suchen. Sie fanden eine subtile Veränderung im Gen COL5A1, das an der Bildung von Typ-V-Kollagen beteiligt ist. Diese Veränderung lag nicht im üblichen protein-kodierenden Bereich des Gens, sondern tief in einem nichtkodierenden Abschnitt. Auf den ersten Blick war eine solche versteckte Veränderung schwer einzuordnen, weshalb die Forscher testen mussten, ob sie tatsächlich die Art und Weise stört, wie die Genbotschaft gelesen wird.
Ein Labortest, der das Genablesen nachahmt
Um die Wirkung dieser verborgenen DNA-Veränderung zu untersuchen, verwendeten die Wissenschaftler ein Werkzeug, das als Minigen-Assay bekannt ist. Sie konstruierten zwei Miniaturversionen des COL5A1-Genabschnitts: eine mit der normalen Sequenz und eine mit der Varianten der Familie. Diese Konstrukte wurden in menschliche Zellen im Labor eingebracht, um zu beobachten, wie die zelluläre Maschinerie sie verarbeiten würde. Die normale Variante erzeugte die erwartete RNA-Botschaft, während die veränderte Variante zwei Botschaften produzierte. Eine wirkte normal, die andere enthielt ein zusätzlich eingelagertes Stück — ein sogenanntes Pseudoexon —, das aus einem eigentlich stillen Abschnitt genetischen Materials herausgespleißt worden war.
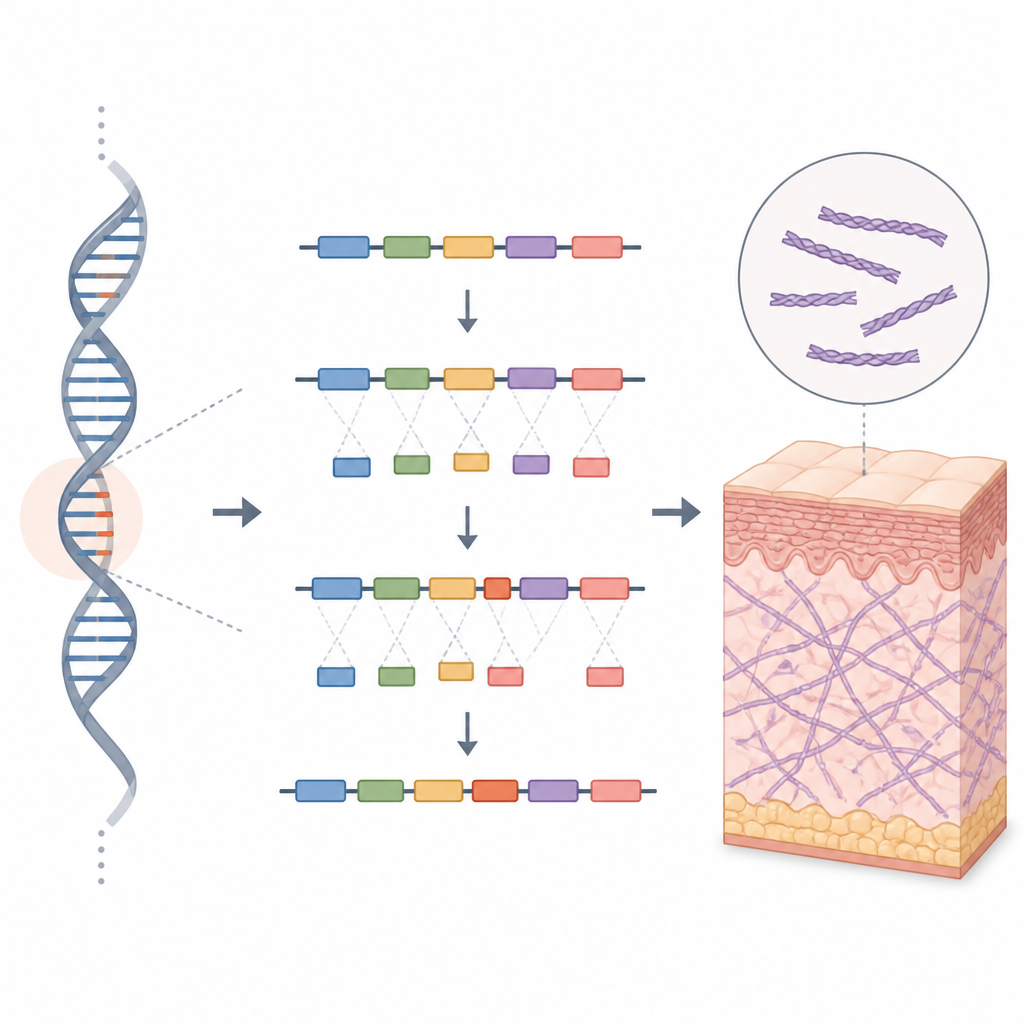
Wie ein winziges zusätzliches Stück Gewebe schwächt
Dieses zusätzlich eingebaute Stück verschob den Leserahmen der Genbotschaft und führte schnell zu einem Stopp-Signal, das das Protein verkürzte. Das Ergebnis wird voraussichtlich entweder ein verkürztes, funktionsloses Fragment des COL5A1-Proteins oder gar kein Protein sein, da Zellen solche fehlerhaften Botschaften oft abbauen. In beiden Fällen stünde dem Körper zu wenig funktionelles Typ-V-Kollagen zur Verfügung, wodurch das Kollagennetz, das Haut und Gelenke stützt, geschwächt würde. Das Muster in der Familie, das Fehlen dieser Variante in Bevölkerungsdatenbanken und die klaren Labornachweise für fehlerhaftes Spleißen führten die Autoren dazu, die Veränderung nach aktuellen Richtlinien der klinischen Genetik als wahrscheinlich krankheitsverursachend einzustufen.
Warum diese versteckte Veränderung wichtig ist
Diese Arbeit zeigt, dass bedeutende krankheitsverursachende Veränderungen weit entfernt von den üblichen, leicht durchsuchbaren Genabschnitten liegen können und dass spezialisierte RNA-Level-Tests nötig sein können, wenn routinemäßige genetische Screenings ein starkes klinisches Bild nicht erklären. Sie deutet auch auf zukünftige Behandlungsoptionen hin, die theoretisch diesen Fehler korrigieren könnten, indem sie das zusätzliche Pseudoexon blockieren und die normale Botschaft wiederherstellen — etwa durch antisense Oligonukleotide. Für Menschen mit klassischem Ehlers-Danlos-Syndrom vertiefen Studien wie diese das Verständnis dafür, warum ihre Gewebe fragil sind, und könnten schließlich den Weg zu präziseren Diagnosen und maßgeschneiderten Therapien ebnen.
Zitation: Zhao, J., Feng, J. A novel deep intronic COL5A1 variant in an Ehlers-Danlos syndrome family: functional characterization by minigene assay. Sci Rep 16, 15232 (2026). https://doi.org/10.1038/s41598-026-46346-8
Schlüsselwörter: Ehlers-Danlos-Syndrom, COL5A1, intronische Variante, aberranter Spleißvorgang, Bindegewebe