Clear Sky Science · it
Una nuova variante intronica profonda di COL5A1 in una famiglia con sindrome di Ehlers-Danlos: caratterizzazione funzionale mediante saggio minigene
Indizi nascosti nella pelle elastica
Perché alcune persone si lividano facilmente, formano cicatrici profonde e hanno pelle insolitamente elastica e articolazioni molto mobili? Per le famiglie che convivono con la sindrome di Ehlers-Danlos, questi problemi quotidiani possono essere dolorosi e fonte di confusione. Questo studio segue una di queste famiglie in Cina e mostra come una singola modifica, ben nascosta nel loro DNA, possa indebolire l’impalcatura interna del corpo, offrendo nuove informazioni per la diagnosi e possibili idee terapeutiche future per questa condizione rara.
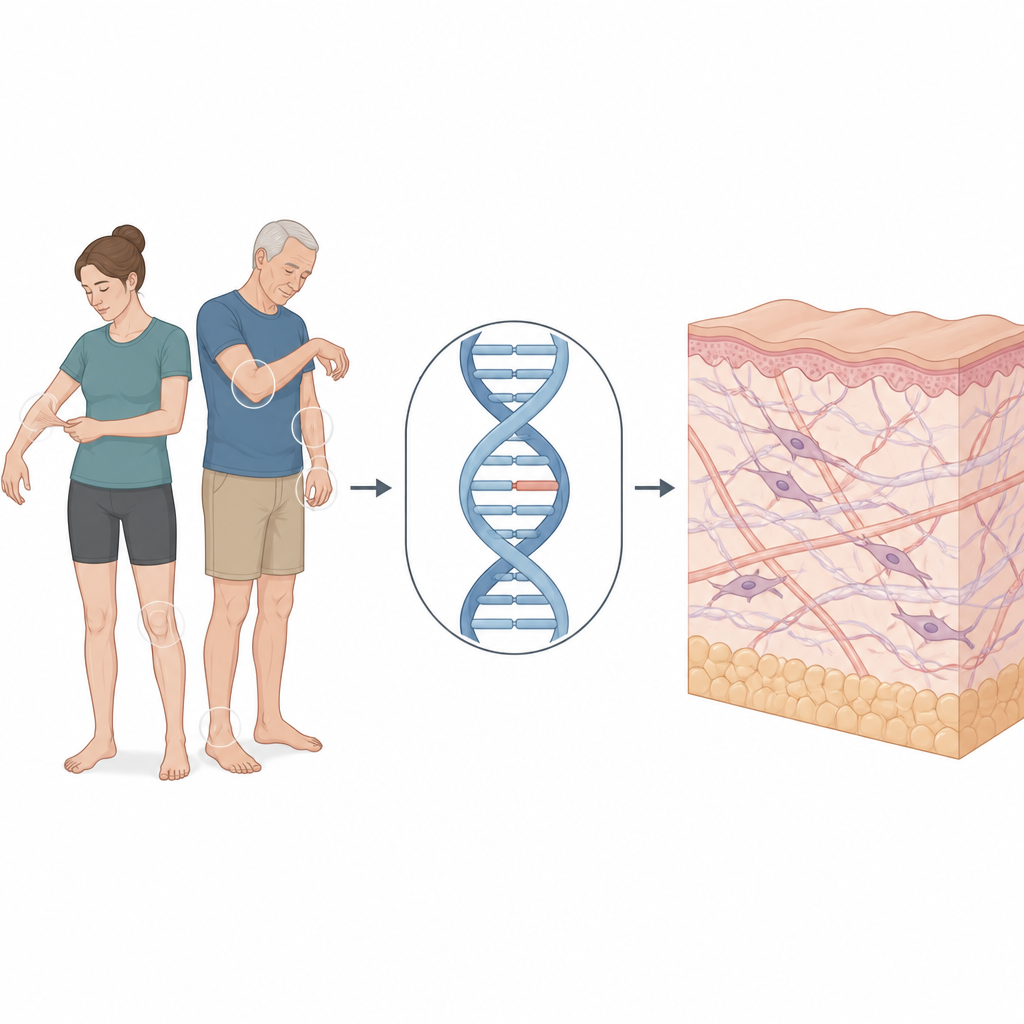
Quello che affrontava questa famiglia
La ricerca si concentra su una donna di 30 anni che si è rivolta a una clinica dermatologica per problemi cronici di pelle e articolazioni. Aveva una pelle morbida e vellutata che si allungava più del normale, cicatrici sottili e carta velina durante la guarigione, e si procurava lividi con poco sforzo. Le articolazioni delle dita si piegavano oltre il normale. Il padre mostrava pelle lassa e alcuni tratti facciali spesso osservati in questa condizione, mentre la madre non presentava sintomi. Nel complesso, i loro segni indirizzavano verso il tipo classico di sindrome di Ehlers-Danlos, una forma che interessa principalmente pelle e articolazioni ma può anche destare preoccupazione per i vasi sanguigni e altri organi.
Cercare risposte nei geni
La sindrome di Ehlers-Danlos classica è di solito collegata a varianti nei geni che costruiscono il collagene, le corde proteiche che rinforzano la pelle e molti altri tessuti. Il team ha usato il sequenziamento dell’esoma clinico, che esamina migliaia di geni associati a malattie, per cercare cambiamenti sospetti nella paziente e nei suoi genitori. Hanno trovato una alterazione sottile in un gene chiamato COL5A1, che contribuisce a produrre il collagene di tipo V. Questa variante non si trovava nella solita porzione codificante della proteina, ma in profondità in un segmento non codificante. A prima vista, una modifica nascosta così era difficile da classificare, quindi i ricercatori hanno dovuto testare se disturbasse realmente il modo in cui il messaggio genico viene letto.
Un test di laboratorio che imita la lettura del gene
Per sondare l’effetto di questa modifica del DNA sepolta, gli scienziati hanno usato uno strumento noto come saggio minigene. Hanno costruito due versioni in miniatura del segmento genico COL5A1: una con la sequenza normale e una con la variante della famiglia. Questi costrutti sono stati inseriti in cellule umane coltivate in laboratorio per vedere come la macchina cellulare li avrebbe processati. La versione normale ha prodotto il messaggio di RNA atteso, mentre la versione alterata ha prodotto due messaggi. Uno sembrava normale, ma l’altro conteneva un pezzo inserito in più, un cosiddetto pseudoesone, ricavato da ciò che avrebbe dovuto essere una porzione silente di materiale genetico.
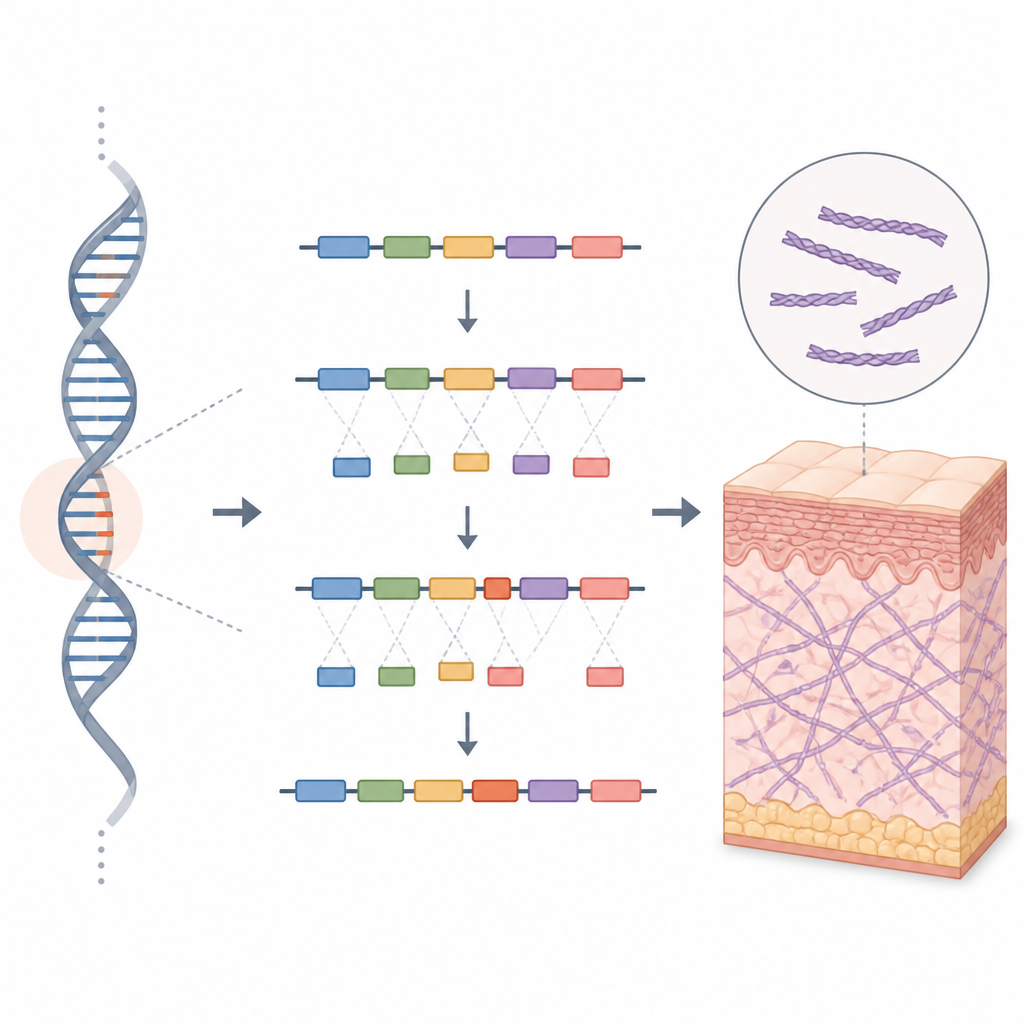
Come un minuscolo pezzo extra causa tessuto fragile
Questo pezzo inserito ha spostato il quadro di lettura del messaggio genico, introducendo rapidamente un segnale di stop che tronca la proteina. Il risultato si prevede sia una frazione tronca e non funzionante della proteina COL5A1 sia l’assenza completa della proteina, poiché le cellule spesso degradano messaggi difettosi di questo tipo. In entrambi i casi, l’organismo avrebbe troppo poco collagene di tipo V funzionale, indebolendo la rete di collagene che sostiene pelle e articolazioni. Il quadro clinico nella famiglia, l’assenza di questa variante nei database di popolazione e la chiara evidenza in laboratorio di uno splicing difettoso hanno portato gli autori a classificare la modifica come verosimilmente patogenica secondo le attuali linee guida di genetica clinica.
Perché questa modifica nascosta conta
Questo lavoro dimostra che varianti rilevanti per la malattia possono nascondersi lontano dalle parti usuali e facilmente esaminabili dei geni, e che possono essere necessari test specializzati a livello di RNA quando gli screening genetici di routine non riescono a spiegare un quadro clinico evidente. Suggerisce inoltre possibili terapie future che, in linea di principio, potrebbero correggere questo tipo di errore bloccando lo pseudoesone in eccesso e ripristinando il messaggio normale, con approcci come gli oligonucleotidi antisenso. Per le persone con la forma classica di Ehlers-Danlos, studi di questo tipo approfondiscono la comprensione del perché i loro tessuti siano fragili e potrebbero infine aprire la strada a diagnosi più precise e terapie su misura.
Citazione: Zhao, J., Feng, J. A novel deep intronic COL5A1 variant in an Ehlers-Danlos syndrome family: functional characterization by minigene assay. Sci Rep 16, 15232 (2026). https://doi.org/10.1038/s41598-026-46346-8
Parole chiave: Sindrome di Ehlers-Danlos, COL5A1, variante intronica, splicing aberrante, tessuto connettivo