Clear Sky Science · it
Un potenziale interatomico di apprendimento automatico a uso generale per il sistema Mg-Al-Si-O adatto ai materiali terrestri in condizioni di alta pressione e temperatura
Uno sguardo al cuore nascosto della Terra
In profondità sotto i nostri piedi, rocce costituite da magnesio, alluminio, silicio e ossigeno modellano tutto, dalle eruzioni vulcaniche alla tettonica a placche. Tuttavia le estreme pressioni e temperature dell’interno terrestre rendono questi materiali quasi impossibili da studiare direttamente in laboratorio. Questo articolo presenta un nuovo metodo basato su computer per riprodurre il comportamento di tali minerali, usando l’intelligenza artificiale moderna per colmare il divario tra calcoli quantistici costosi e modelli tradizionali eccessivamente semplificati.
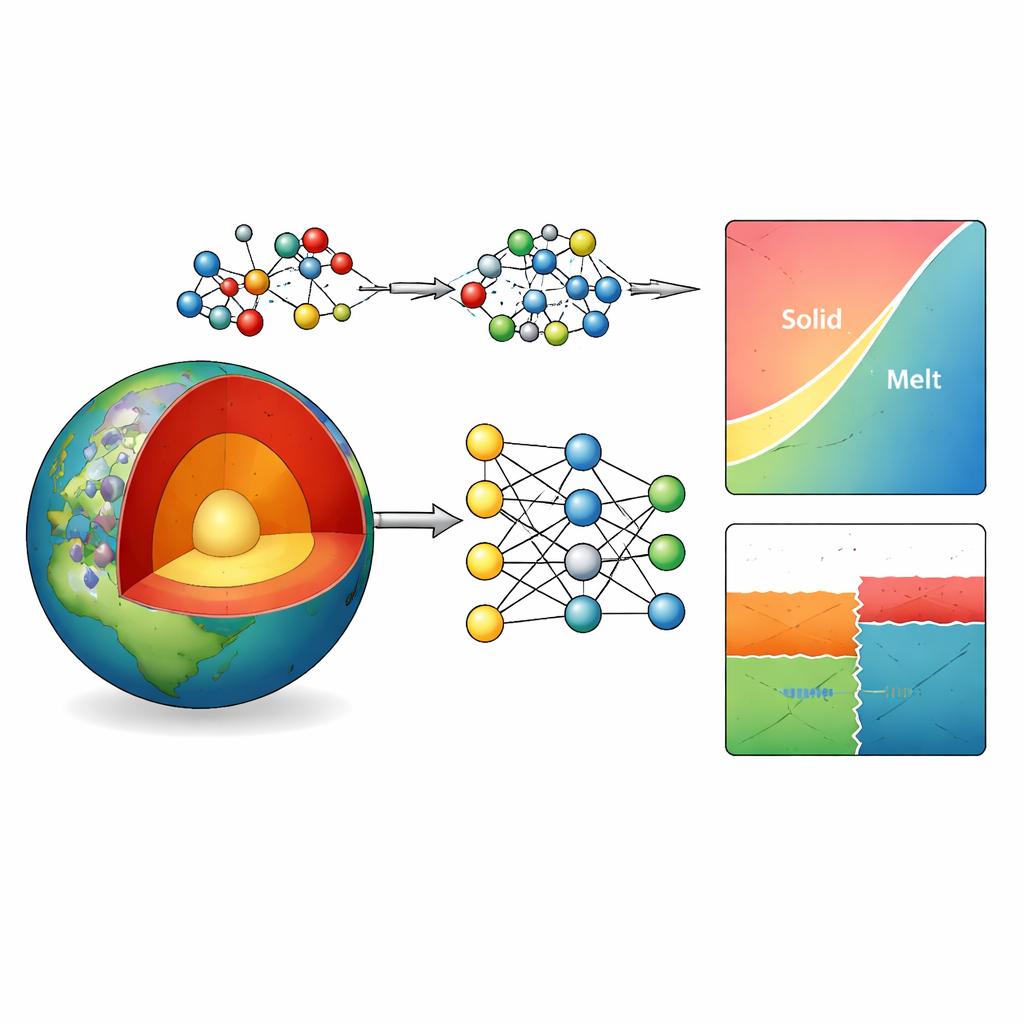
Perché simulare le rocce profonde è così difficile
Il mantello terrestre è dominato da minerali formati da magnesio, alluminio, silicio e ossigeno. La loro stabilità e il comportamento di fusione controllano quanto le rocce siano rigide o fluide, come le placche sprofondano nel mantello e dove si verificano terremoti e discontinuità di velocità sismica. I ricercatori si affidano ai diagrammi di fase—mappe che mostrano quali minerali sono stabili a una data profondità e temperatura—per capire questi processi. Gli esperimenti a profondità di centinaia di chilometri sono estremamente impegnativi, quindi gli scienziati ricorrono alle simulazioni al calcolatore. I modelli classici sono veloci ma spesso sbagliano le transizioni mineralogiche, mentre i metodi quantistici più accurati sono troppo lenti per i sistemi grandi e complessi di interesse per la geoscienza.
Insegnare a una rete neurale le leggi degli atomi
Gli autori risolvono questo collo di bottiglia costruendo un “potenziale interatomico” basato sull’apprendimento automatico per il sistema Mg–Al–Si–O. In pratica, addestrano una rete neurale profonda a prevedere come gli atomi si attraggono e si respingono, basandosi su un ampio archivio di strutture di esempio. Questi esempi sono generati mediante calcoli quantistici avanzati con un metodo particolarmente affidabile (chiamato r2SCAN) su un’ampia gamma di pressioni, temperature e tipi di minerali e fuso. Per affinare ulteriormente la precisione proprio dove conta—piccole differenze di energia che determinano quale minerale è stabile—aggiungono una semplice correzione gaussiana a coppie tarata su un database termodinamico affidabile. Questo approccio ibrido riduce l’errore energetico medio per 20 minerali comuni del mantello da circa 5 a poco più di 1 kilojoule per mole, senza alterare in modo significativo i loro volumi.
Verificare il modello rispetto alle mappe di fase naturali
Armati di questo potenziale raffinato, il team calcola diagrammi di fase per sistemi chiave, inclusi la silice pura, gli alluminosilicati e le composizioni di silicato di magnesio che comprendono minerali principali del mantello come la forsterite, la wadsleyite e la ringwoodite. Utilizzando l’integrazione termodinamica e relazioni standard della chimica fisica, tracciano i confini in cui una fase cede il posto a un’altra o al fuso. I diagrammi previsti corrispondono strettamente ai risultati sperimentali, cogliendo persino transizioni ad alta pressione nella silice che non erano state incluse esplicitamente nei dati di addestramento. Dove emergono differenze—come uno spostamento modesto del confine tra forsterite e wadsleyite—possono essere ricondotte a incertezze energetiche residue di solo pochi kilojoule per mole, equivalenti a errori di alcune centinaia di gradi o a una frazione di gigapascal.
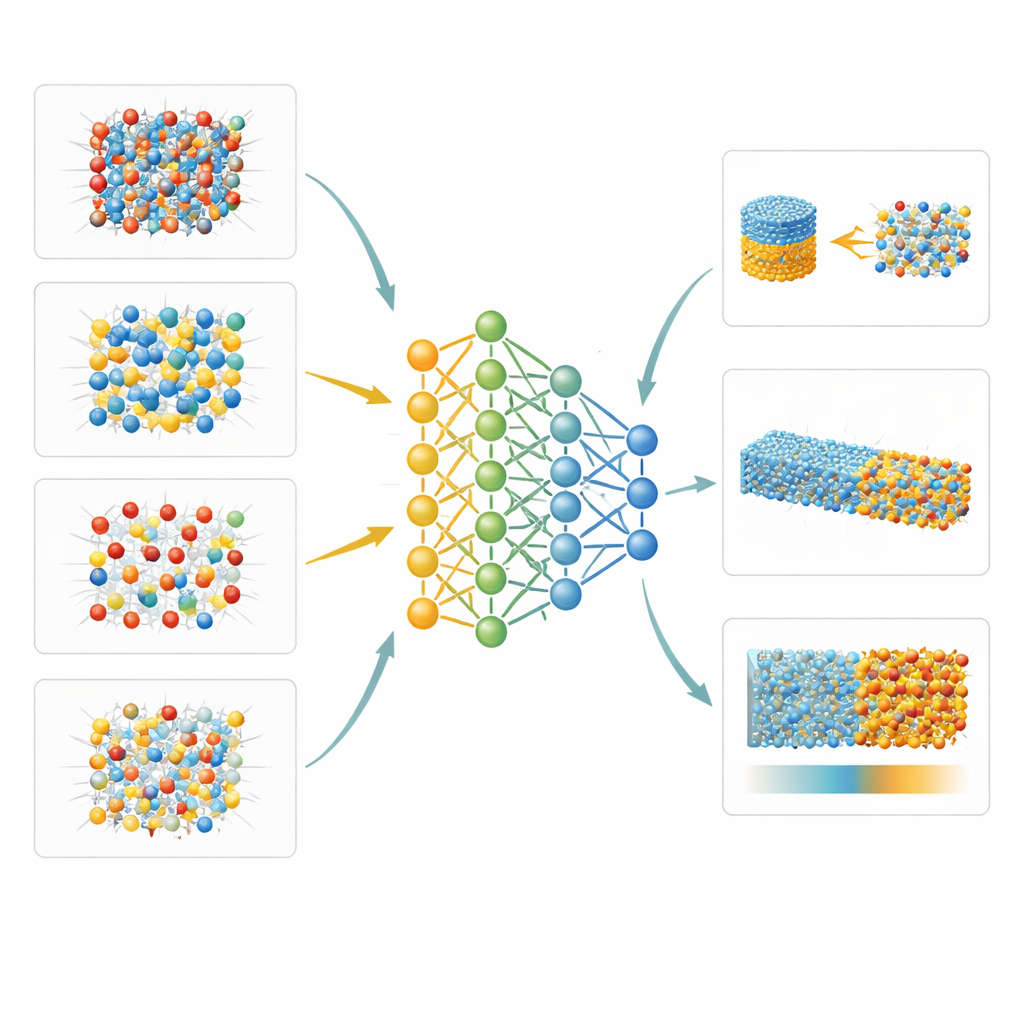
Nuove visioni di interfacce e disordine atomico
Poiché il modello appreso è sia accurato sia sufficientemente veloce per sistemi di grandi dimensioni, gli autori possono esplorare proprietà quasi inaccessibili in laboratorio. Un esempio è il modo in cui gli atomi di alluminio e silicio si riorganizzano all’interno del minerale sillimanite con l’aumento della temperatura, studiato combinando il loro potenziale con simulazioni Monte Carlo. Un altro è l’energia libera del confine tra cristalli solidi e roccia fusa per due minerali rappresentativi: la periclase (MgO) e la forsterite (una forma di olivina). Usando metodi di campionamento sofisticati, mostrano che queste interfacce presentano un’anisotropia relativamente bassa—la differenza di energia superficiale tra facce cristalline—circa 6 percento per la periclase e 12 percento per la forsterite. Indagano inoltre come lo stress non uniforme influenzi la nota trasformazione del quarzo a bassa temperatura nella sua forma ad alta temperatura, rilevando che livelli geologici tipici di stress differenziale spostano la transizione solo marginalmente.
Cosa significa per la comprensione del nostro pianeta
Per un non specialista, il risultato principale è che gli autori hanno creato un potente nuovo “laboratorio digitale” per i materiali della Terra profonda. Il loro modello di apprendimento automatico può riprodurre confini mineralogici noti e poi andare oltre gli esperimenti attuali per stimare proprietà sottili di fusi, interfacce e disordine atomico in condizioni estreme. Questo apre la strada a simulazioni più realistiche del flusso del mantello, della fusione e delle strutture sismiche, aiutando gli scienziati a collegare il comportamento degli atomi con i processi su larga scala dell’interno del nostro pianeta.
Citazione: Zhong, X., Li, Y. & John, T. A general purposed machine learning interatomic potential for Mg-Al-Si-O system suitable for Earth materials at high pressure and temperature conditions. npj Comput Mater 12, 141 (2026). https://doi.org/10.1038/s41524-026-02056-3
Parole chiave: Materiali del mantello terrestre, potenziale di apprendimento automatico, dinamica molecolare, diagrammi di fase, interfacce solido–fuso