Clear Sky Science · fr
Un potentiel interatomique par apprentissage automatique à usage général pour le système Mg-Al-Si-O adapté aux matériaux terrestres aux hautes pressions et températures
Observer le cœur caché de la Terre
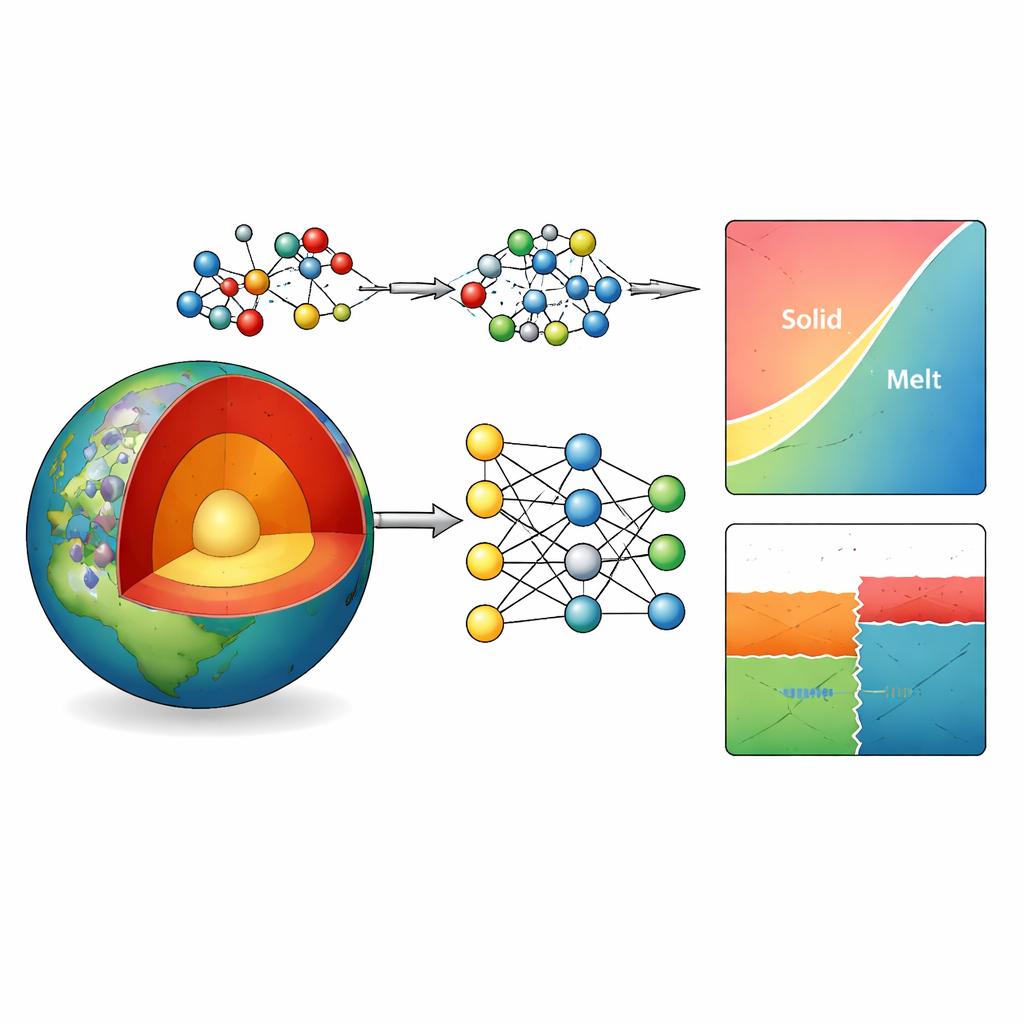
Au plus profond de la Terre, des roches composées de magnésium, d’aluminium, de silicium et d’oxygène contrôlent tout, des éruptions volcaniques à la tectonique des plaques. Pourtant, les pressions et températures extrêmes de l’intérieur terrestre rendent ces matériaux presque impossibles à étudier directement en laboratoire. Cet article présente une nouvelle méthode informatique pour reproduire le comportement de ces minéraux, utilisant l’intelligence artificielle moderne pour faire le lien entre des calculs quantiques coûteux et des modèles traditionnels trop simplifiés.
Pourquoi il est si difficile de simuler les roches profondes
Le manteau terrestre est dominé par des minéraux formés de magnésium, d’aluminium, de silicium et d’oxygène. Leur stabilité et leur comportement de fusion déterminent la viscosité des roches, la façon dont les plaques plongent dans le manteau et où se produisent les séismes et les sauts de vitesse sismique. Les chercheurs s’appuient sur des diagrammes de phases — cartes montrant quels minéraux sont stables à une profondeur et une température données — pour comprendre ces processus. Les expériences reproduisant des conditions à des centaines de kilomètres de profondeur sont extrêmement difficiles, aussi les scientifiques se tournent-ils vers la simulation numérique. Les modèles classiques sont rapides mais se trompent souvent sur les transitions minérales, tandis que les méthodes quantiques plus précises sont beaucoup trop lentes pour les grands systèmes complexes qui intéressent les géoscientifiques.
Apprendre à un réseau neuronal les lois des atomes
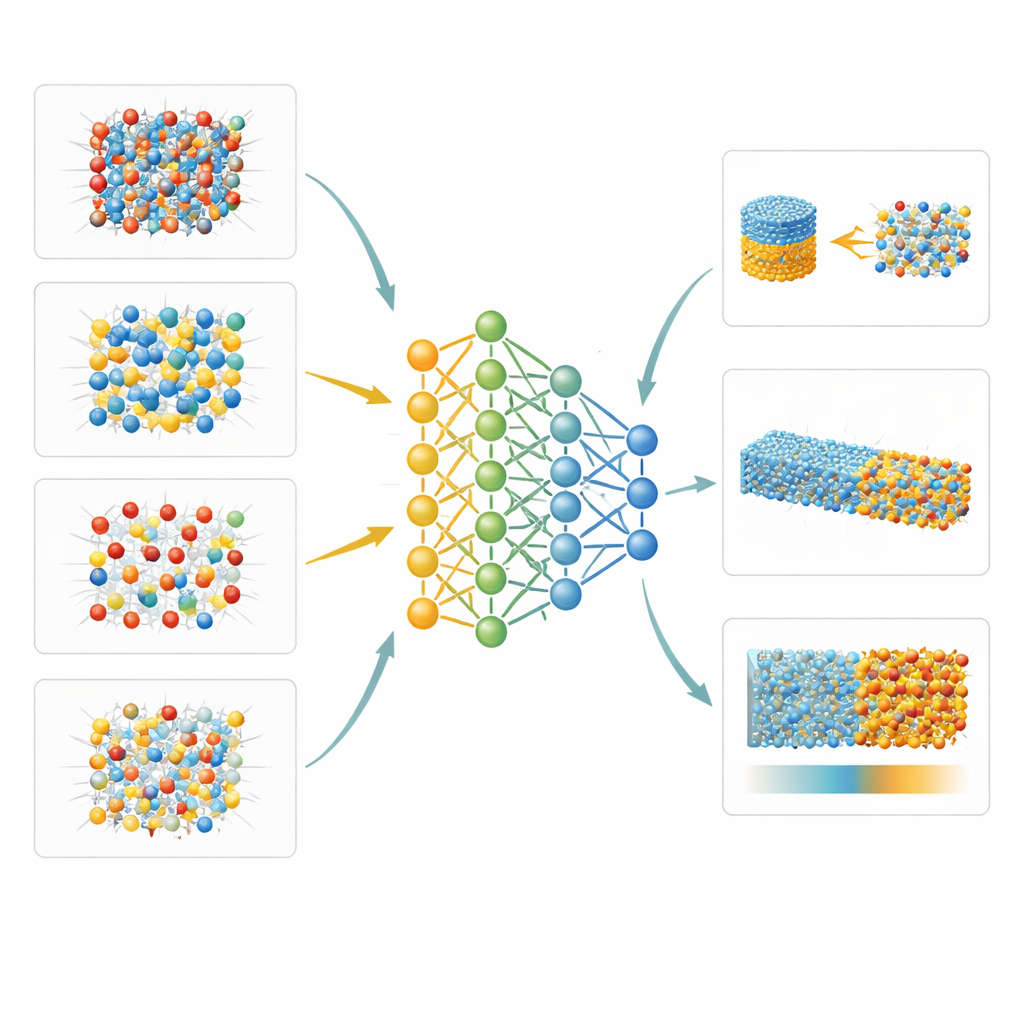
Les auteurs s’attaquent à ce goulot d’étranglement en construisant un « potentiel interatomique » par apprentissage automatique pour le système Mg–Al–Si–O. Essentiellement, ils entraînent un réseau neuronal profond à prédire comment les atomes s’attirent et se repoussent, à partir d’une grande bibliothèque de structures exemples. Ces exemples sont générés par des calculs quantiques avancés avec une méthode particulièrement fiable (appelée r2SCAN) sur une vaste gamme de pressions, températures et types de minéraux et de phases fondues. Pour améliorer encore la précision là où cela compte le plus — de minuscules différences d’énergie qui déterminent quel minéral est stable — ils ajoutent une correction gaussienne simple en paires, ajustée sur une base de données thermodynamique de référence. Cette approche hybride réduit l’erreur énergétique moyenne pour 20 minéraux courants du manteau, d’environ 5 à un peu plus de 1 kilojoule par mole, sans perturber significativement leurs volumes.
Vérifier le modèle par rapport aux cartes de phases naturelles
Avec ce potentiel affiné, l’équipe calcule des diagrammes de phases pour des systèmes clés, notamment la silice pure, les aluminosilicates et des compositions silicatées de magnésium couvrant des minéraux majeurs du manteau comme la forstérite, la wadsleyite et la ringwoodite. En utilisant l’intégration thermodynamique et des relations standard de la chimie physique, ils tracent les frontières où une phase laisse place à une autre ou au liquide. Les diagrammes prédits correspondent étroitement aux résultats expérimentaux, capturant même des transitions à haute pression de la silice qui n’étaient pas explicitement incluses dans les données d’entraînement. Là où existent des différences — par exemple un léger décalage dans la frontière entre forstérite et wadsleyite — elles s’expliquent par des incertitudes énergétiques restantes de seulement quelques kilojoules par mole, équivalant à des erreurs de quelques centaines de degrés ou d’une fraction de gigapascal.
Nouvelles perspectives sur les interfaces et le désordre atomique
Parce que le modèle appris par machine est à la fois précis et suffisamment rapide pour de grands systèmes, les auteurs peuvent explorer des propriétés quasi inaccessibles en laboratoire. Un exemple est la manière dont les atomes d’aluminium et de silicium se réarrangent dans le minéral sillimanite quand la température augmente, qu’ils étudient en combinant leur potentiel avec des simulations de Monte Carlo. Un autre exemple est l’énergie libre de la frontière entre cristaux solides et roche fondue pour deux minéraux représentatifs : la périclase (MgO) et la forstérite (une forme d’olivine). En utilisant des méthodes d’échantillonnage sophistiquées, ils montrent que ces interfaces présentent une anisotropie relativement faible — la différence d’énergie de surface entre faces cristallines — d’environ 6 % pour la périclase et 12 % pour la forstérite. Ils examinent aussi comment un état de contrainte inégal affecte la transformation bien connue du quartz basse température vers sa forme haute température, trouvant que des niveaux géologiques typiques de contrainte différentielle ne déplacent la transition que légèrement.
Ce que cela signifie pour la compréhension de notre planète
Pour un non-spécialiste, l’essentiel est que les auteurs ont créé un « laboratoire numérique » puissant pour les matériaux du manteau profond. Leur modèle d’apprentissage automatique peut reproduire les frontières minérales connues puis aller au-delà des expériences actuelles pour estimer des propriétés subtiles des phases fondues, des interfaces et du désordre atomique sous conditions extrêmes. Cela ouvre la porte à des simulations plus réalistes de l’écoulement du manteau, de la fusion et des structures sismiques, aidant les scientifiques à relier le comportement des atomes aux mécanismes à grande échelle de l’intérieur de notre planète.
Citation: Zhong, X., Li, Y. & John, T. A general purposed machine learning interatomic potential for Mg-Al-Si-O system suitable for Earth materials at high pressure and temperature conditions. npj Comput Mater 12, 141 (2026). https://doi.org/10.1038/s41524-026-02056-3
Mots-clés: Matériaux du manteau terrestre, potentiel d’apprentissage automatique, dynamique moléculaire, diagrammes de phases, interfaces solide–fusion