Clear Sky Science · es
Un potencial interatómico de propósito general basado en aprendizaje automático para el sistema Mg-Al-Si-O adecuado para materiales terrestres en condiciones de alta presión y temperatura
Mirando al corazón oculto de la Tierra
En lo profundo bajo nuestros pies, las rocas compuestas de magnesio, aluminio, silicio y oxígeno controlan desde las erupciones volcánicas hasta la tectónica de placas. Sin embargo, las presiones y temperaturas extremas del interior terrestre hacen que estos materiales sean casi imposibles de estudiar directamente en el laboratorio. Este artículo presenta una nueva forma computacional de emular el comportamiento de estos minerales, empleando la inteligencia artificial moderna para salvar la brecha entre costosos cálculos cuánticos y modelos tradicionales demasiado simplificados.
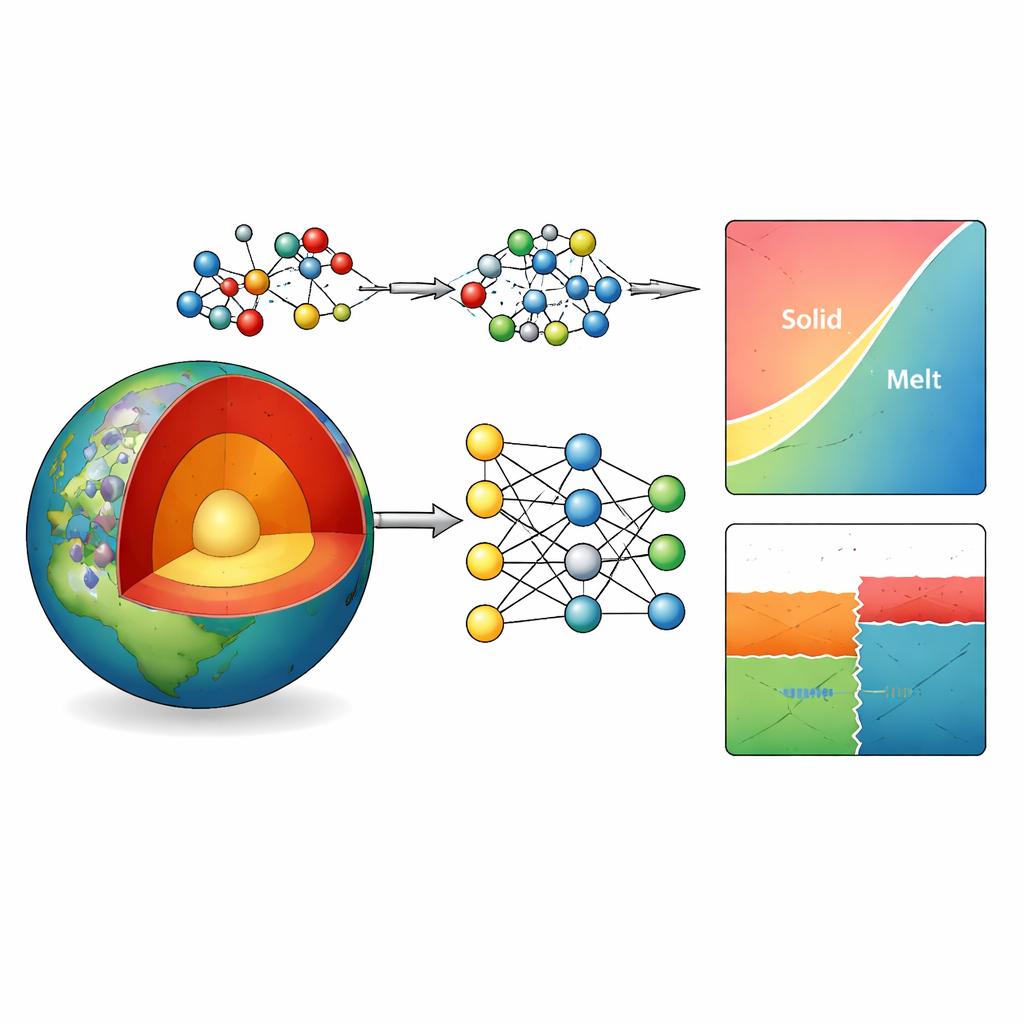
Por qué es tan difícil simular las rocas profundas
El manto terrestre está dominado por minerales formados por magnesio, aluminio, silicio y oxígeno. Su estabilidad y comportamiento de fusión controlan cuán rígidas o fluidas son las rocas, cómo las placas se hunden en el manto y dónde ocurren terremotos y saltos en la velocidad sísmica. Los investigadores dependen de diagramas de fases—mapas que muestran qué minerales son estables a una profundidad y temperatura dadas—para entender estos procesos. Realizar experimentos a cientos de kilómetros de profundidad es extremadamente desafiante, por lo que los científicos recurren a simulaciones por ordenador. Los modelos clásicos son rápidos pero con frecuencia predicen mal las transiciones minerales, mientras que los métodos cuántico‑mecánicos más precisos son demasiado lentos para los sistemas grandes y complejos que interesan a los geocientíficos.
Enseñando a una red neuronal las reglas de los átomos
Los autores abordan este cuello de botella construyendo un "potencial interatómico" basado en aprendizaje automático para el sistema Mg–Al–Si–O. En esencia, entrenan una red neuronal profunda para predecir cómo los átomos se atraen y repelen entre sí, a partir de una amplia biblioteca de estructuras de ejemplo. Estos ejemplos se generan mediante cálculos cuánticos avanzados con un método particularmente fiable (denominado r2SCAN) a lo largo de un amplio rango de presiones, temperaturas y tipos de minerales y fundidos. Para afinar la precisión aún más donde más importa—pequeñas diferencias de energía que deciden qué mineral es estable—añaden una corrección gaussiana pareada sencilla ajustada contra una base de datos termodinámica de confianza. Este enfoque híbrido reduce el error energético medio para 20 minerales comunes del manto de aproximadamente 5 a poco más de 1 kilojulio por mol, sin alterar de forma significativa sus volúmenes.
Comprobando el modelo frente a los mapas de fases naturales
Con este potencial refinado, el equipo calcula diagramas de fases para sistemas clave, incluyendo sílice pura, aluminosilicatos y composiciones de silicatos de magnesio que abarcan minerales principales del manto como forsterita, wadsleyita y ringwoodita. Usando integración termodinámica y relaciones estándar de la química física, trazan los límites donde una fase da paso a otra o a la fusión. Los diagramas predichos concuerdan de cerca con los resultados experimentales, incluso capturando transiciones de alta presión en la sílice que no se incluyeron explícitamente en los datos de entrenamiento. Donde hay diferencias—como un desplazamiento moderado en la frontera entre forsterita y wadsleyita—pueden atribuirse a incertidumbres energéticas remanentes de solo unos pocos kilojulios por mol, equivalentes a errores de unas pocas centenas de grados o a una fracción de gigapascal.
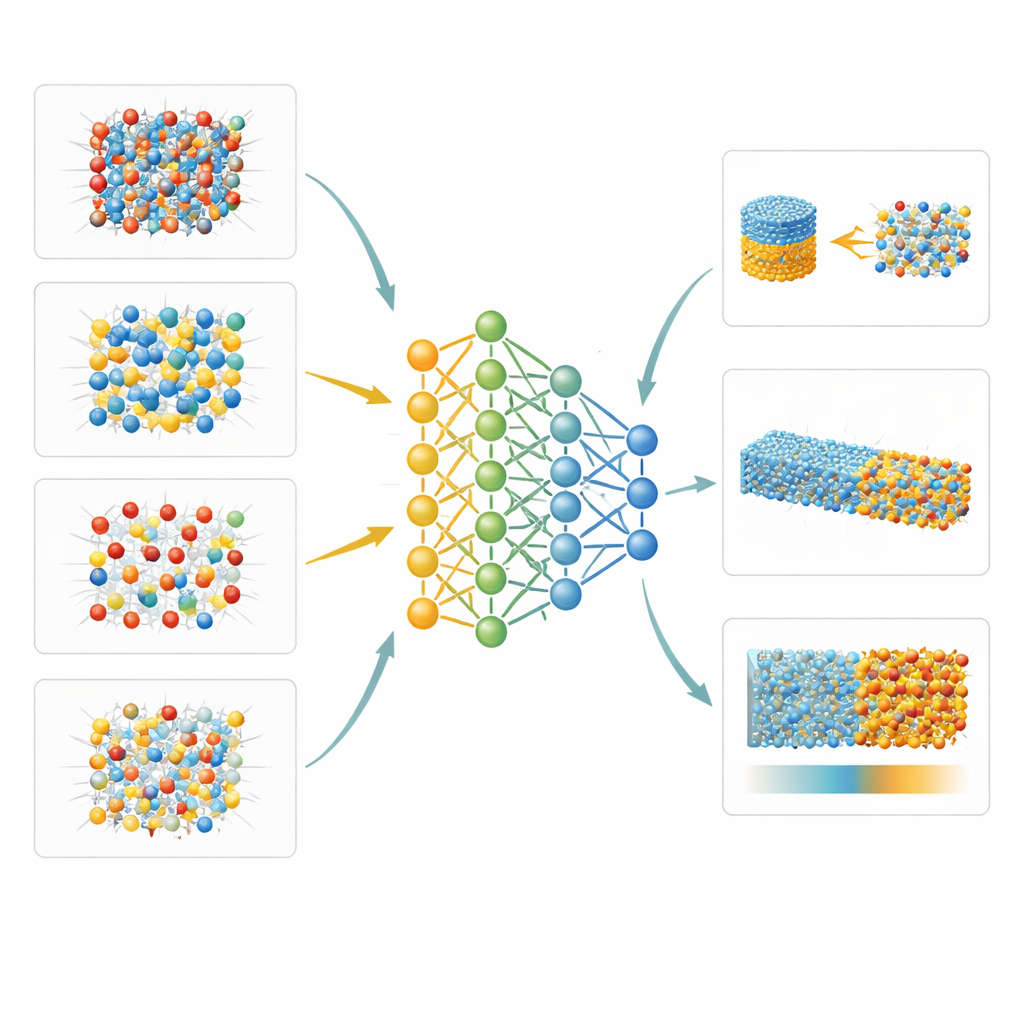
Nuevas perspectivas sobre interfaces y desorden atómico
Porque el modelo aprendido por la máquina es a la vez preciso y lo bastante rápido para sistemas grandes, los autores pueden explorar propiedades casi inaccesibles en el laboratorio. Un ejemplo es la manera en que los átomos de aluminio y silicio se reorganizan dentro del mineral silimanita al aumentar la temperatura, que estudian combinando su potencial con simulaciones de Monte Carlo. Otro es la energía libre de la frontera entre cristales sólidos y roca fundida para dos minerales representativos: periclasa (MgO) y forsterita (una forma de olivino). Empleando métodos de muestreo sofisticados, muestran que estas interfaces tienen una anisotropía relativamente baja—la diferencia en energía superficial entre caras cristalinas—alrededor del 6 por ciento para periclasa y del 12 por ciento para forsterita. También investigan cómo el esfuerzo desigual afecta la conocida transformación del cuarzo de baja temperatura a su forma de alta temperatura, encontrando que los niveles geológicos típicos de esfuerzo diferencial desplazan la transición solo ligeramente.
Qué significa esto para entender nuestro planeta
Para un público general, la conclusión es que los autores han creado un poderoso "laboratorio digital" nuevo para materiales del interior de la Tierra. Su modelo de aprendizaje automático puede reproducir límites minerales conocidos y luego ir más allá de los experimentos actuales para estimar propiedades sutiles de fundidos, interfaces y desorden atómico bajo condiciones extremas. Esto abre la puerta a simulaciones más realistas del flujo del manto, la fusión y las estructuras sísmicas, ayudando a los científicos a conectar el comportamiento de los átomos con el funcionamiento a gran escala del interior de nuestro planeta.
Cita: Zhong, X., Li, Y. & John, T. A general purposed machine learning interatomic potential for Mg-Al-Si-O system suitable for Earth materials at high pressure and temperature conditions. npj Comput Mater 12, 141 (2026). https://doi.org/10.1038/s41524-026-02056-3
Palabras clave: Materiales del manto terrestre, potencial de aprendizaje automático, dinámica molecular, diagramas de fases, interfaces sólido–fusión