Clear Sky Science · fr
Une variante de la rétinite pigmentaire associée à PRPF8 induit des défauts autonomes des photorécepteurs dans la rétine neuronale humaine
Pourquoi c’est important pour la vision
La rétinite pigmentaire est une cause majeure de cécité héréditaire, et pour de nombreux patients nous ne comprenons pas encore complètement pourquoi leurs cellules photoréceptrices meurent. Cette étude utilise de petites rétines humaines cultivées en laboratoire pour étudier une forme énigmatique de la maladie causée par un changement subtil dans un gène appelé PRPF8. En recréant la condition en culture, les chercheurs montrent que les dommages peuvent naître au sein même de la rétine neuronale — en particulier dans les cellules détectrices de lumière — plutôt que d’être déclenchés uniquement par les cellules de soutien situées au fond de l’œil. Leur travail révèle aussi des altérations précoces de l’ARN qui pourraient un jour aider au diagnostic ou au suivi de ce trouble à progression lente.
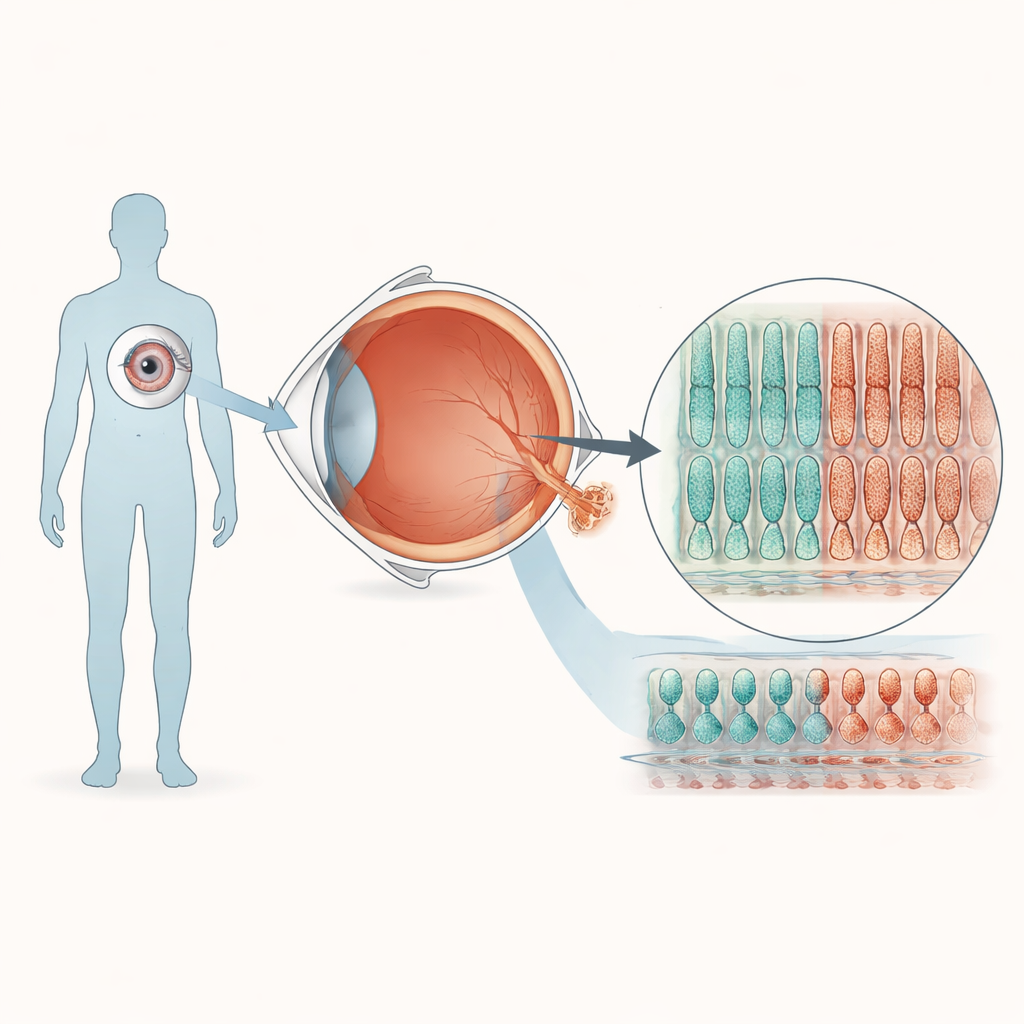
Construire de petites rétines humaines en laboratoire
Pour explorer comment un changement d’une seule lettre dans PRPF8 affecte la vision, l’équipe a transformé des cellules sanguines d’un donneur sain en cellules souches pluripotentes induites, capables d’être orientées vers de nombreux tissus. À l’aide de l’édition génique CRISPR, ils ont créé une lignée cellulaire portant la mutation PRPF8 observée dans la rétinite pigmentaire et une lignée témoin correspondante sans la mutation. Les deux lignées ont ensuite suivi une recette de plusieurs mois pour s’auto-organiser en organoïdes rétiniens tridimensionnels qui reproduisent de nombreuses caractéristiques d’une rétine humaine. Au microscope, les organoïdes issus des cellules mutantes et témoins ont développé des structures en couches similaires et contenaient les principaux types cellulaires rétiniens, ce qui suggère que le développement précoce de l’œil se déroule en grande partie normalement malgré la mutation.
Une faiblesse cachée dans les cellules détectrices de lumière
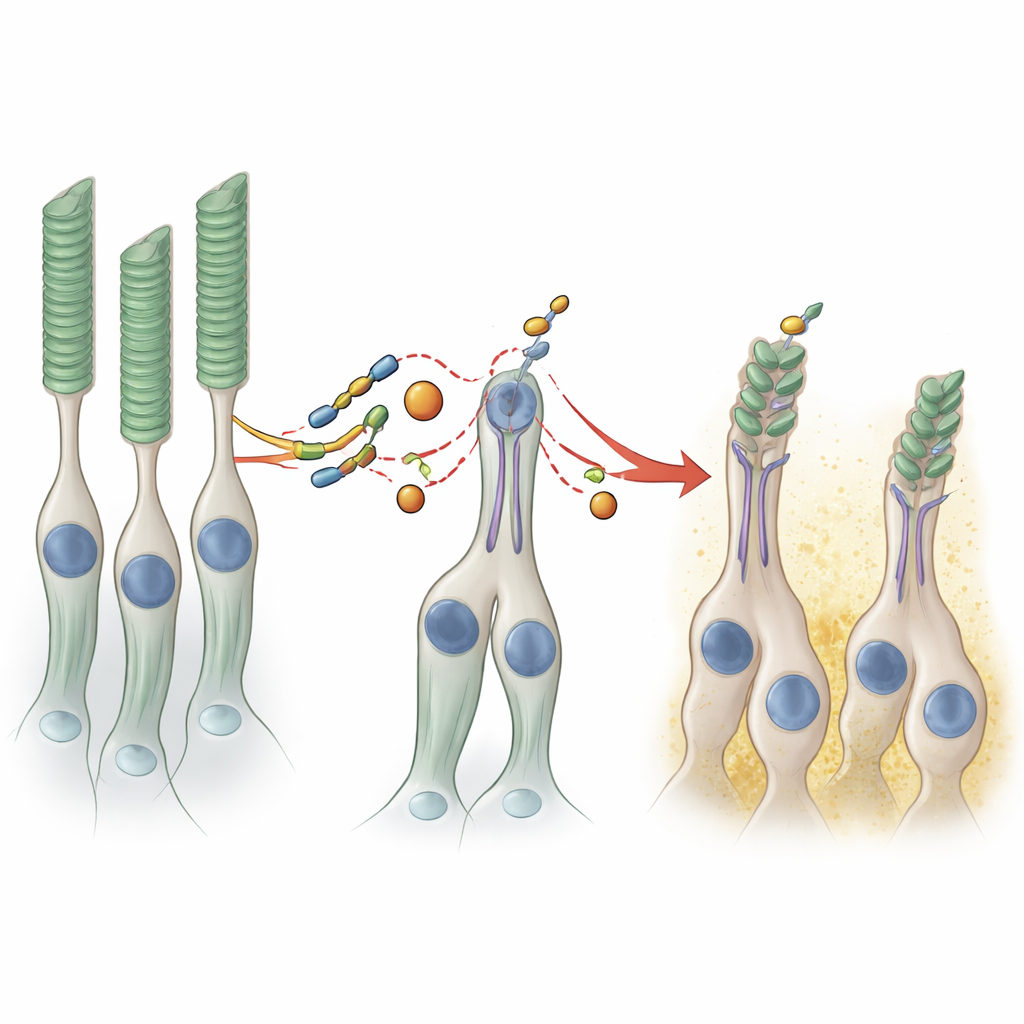
Un examen plus attentif a toutefois révélé que les organoïdes portant la mutation PRPF8 produisaient moins de plusieurs protéines emblématiques des photorécepteurs, les cellules qui captent la lumière. Lorsque les organoïdes ont été laissés à mûrir pendant plus de huit mois, la microscopie électronique et optique a montré une différence structurelle frappante. Dans les organoïdes témoins, la surface était densément couverte de projections ressemblant à des segments internes et externes, les compartiments spécialisés où les photorécepteurs stockent leurs pigments visuels. Dans les organoïdes mutants, ces structures étaient plus clairsemées, et une analyse d’images basée sur l’apprentissage automatique a révélé que la « bordure en brosse » formée par les segments des photorécepteurs était environ deux fois moins épaisse. Parce que ce système contient presque pas d’épithélium pigmentaire rétinien et que les deux tissus sont physiquement séparés, ces défauts témoignent d’une fragilité intrinsèque des photorécepteurs eux-mêmes dans cette forme de la maladie.
Des décalages subtils dans le traitement de l’ARN, de grands effets sur la structure
PRPF8 participe au fonctionnement du spliceosome, la machinerie cellulaire qui édite les messages ARN fraîchement synthétisés. Les chercheurs ont séquencé l’ARN des organoïdes pour voir comment la mutation perturbe ce processus. De façon surprenante, le profil global d’activité génique était seulement modérément modifié ; seulement deux gènes étaient de façon constante plus actifs dans le tissu mutant, et ceux-ci étaient liés aux cellules de soutien telles que les cellules de Müller et les cellules bipolaires. Les changements les plus révélateurs concernaient la façon dont certains ARN étaient épissés. Des centaines d’introns — les segments non codants généralement éliminés — étaient retenus à des taux légèrement plus élevés dans les organoïdes mutants, en particulier ceux présentant des « sites d’épissage » plus faibles. Une poignée d’exons étaient sautés ou inclus plus souvent. Notamment, un gène touché, IFT122, fait partie de la machinerie de transport qui convoie des protéines clés le long du petit pont entre les segments interne et externe des photorécepteurs. Des défauts dans ce système de transport sont déjà connus pour provoquer une dégénérescence rétinienne, ce qui suggère que même de petites erreurs d’épissage dans de tels gènes peuvent compromettre les longs et délicats segments externes.
Les ARN circulaires comme signaux d’alerte précoces
L’équipe a également examiné les ARN circulaires, une classe d’ARN stable mais encore mystérieuse produite lorsque les extrémités de l’ARN se rejoignent pour former une boucle. Dans les organoïdes mutants, plus d’une centaine d’ARN circulaires ont vu leur abondance modifiée par rapport aux témoins, certains augmentant avec l’âge des organoïdes. En comparant ces profils avec un modèle murin portant la même mutation, les chercheurs ont trouvé des gènes hôtes communs produisant des ARN circulaires mal régulés chez les deux espèces. Parce que les ARN circulaires sont de longue durée de vie et s’accumulent avec le temps, leurs niveaux altérés pourraient servir de marqueurs sensibles d’un épissage perturbé et de stress rétinien précoce avant qu’une perte cellulaire massive n’intervienne.
Ce que cela signifie pour les personnes atteintes de rétinite pigmentaire
Dans l’ensemble, ces résultats montrent qu’une mutation apparemment légère de PRPF8 peut affaiblir discrètement les segments externes des photorécepteurs dans un tissu rétinien de type humain, même lorsque les changements d’expression génique globaux sont subtils. Le travail renforce l’idée que, chez au moins certains patients, la maladie débute au sein des cellules détectrices de lumière elles-mêmes, provoquée par des erreurs d’épissage précises dans un nombre restreint de gènes vulnérables impliqués dans les cils et le transport des protéines. Parallèlement, les variations des niveaux d’ARN circulaires émergent comme une signature partagée de ces formes de rétinite pigmentaire liées à l’épissage. À l’avenir, identifier les cibles clés mal épissées et suivre des marqueurs d’ARN circulaires pourrait orienter de nouvelles thérapies visant à corriger le traitement de l’ARN et à préserver la vision.
Citation: Zimmann, F., Banik, P., Kubovčiak, J. et al. PRPF8-associated retinitis pigmentosa variant induces human neural retina-autonomous photoreceptor defects. Sci Rep 16, 10264 (2026). https://doi.org/10.1038/s41598-026-40376-y
Mots-clés: rétinite pigmentaire, dégénérescence des photorécepteurs, épissage de l'ARN, organoïdes rétiniens, ARN circulaire