Clear Sky Science · en
PRPF8-associated retinitis pigmentosa variant induces human neural retina-autonomous photoreceptor defects
Why this matters for eyesight
Retinitis pigmentosa is a leading cause of inherited blindness, yet for many patients we still do not fully understand why their light-sensing cells die. This study uses miniature lab-grown human retinas to probe one puzzling form of the disease caused by a subtle change in a gene called PRPF8. By recreating the condition in a dish, the researchers show that the damage can arise within the neural retina itself—especially in the light-detecting cells—rather than being triggered only by supporting cells at the back of the eye. Their work also reveals early RNA changes that could one day help diagnose or track this slow-moving disorder.
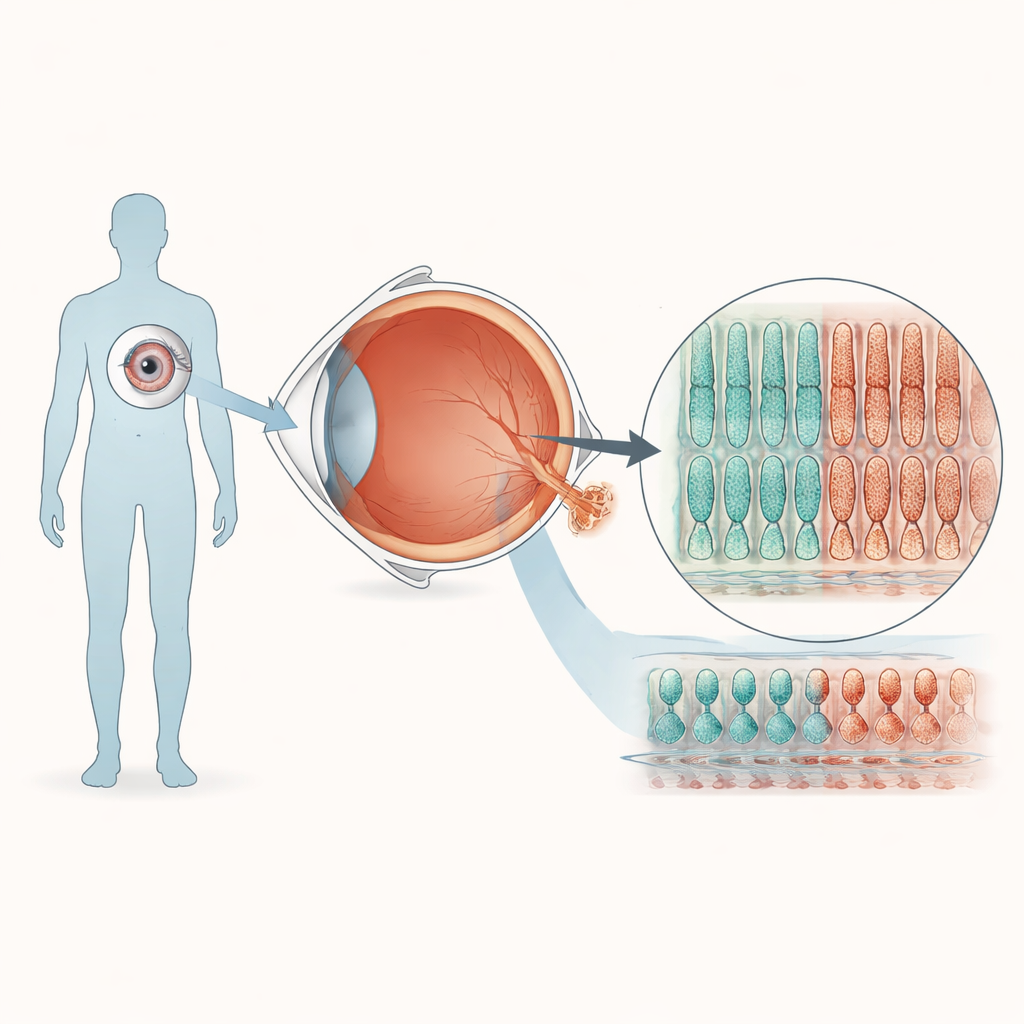
Building tiny human retinas in the lab
To explore how a single-letter change in PRPF8 affects vision, the team turned blood cells from a healthy donor into induced pluripotent stem cells, which can be coaxed to form many tissues. Using CRISPR gene editing, they created one cell line carrying the PRPF8 mutation seen in retinitis pigmentosa and a matching control line without the change. Both lines were then guided through a months-long recipe to self-organize into three-dimensional retinal “organoids” that mimic many features of a human retina. Under the microscope, organoids from mutant and control cells developed similar layered structures and contained the main retinal cell types, suggesting that early eye development proceeds largely normally despite the mutation.
Hidden weakness in the light-sensing cells
Closer inspection, however, revealed that organoids with the PRPF8 mutation produced less of several proteins that are hallmarks of photoreceptors, the cells that capture light. When the organoids were allowed to mature for more than eight months, electron and light microscopy showed a striking structural difference. In control organoids, the surface was densely covered with inner and outer segment–like projections, the specialized compartments where photoreceptors pack their visual pigments. In the mutant organoids, these structures were sparser, and a machine-learning–based image analysis found that the “brush border” formed by photoreceptor segments was about half as thick. Because this system contains almost no retinal pigment epithelium and the two tissues are physically separated, these defects point to an intrinsic fragility of photoreceptors themselves in this form of the disease.
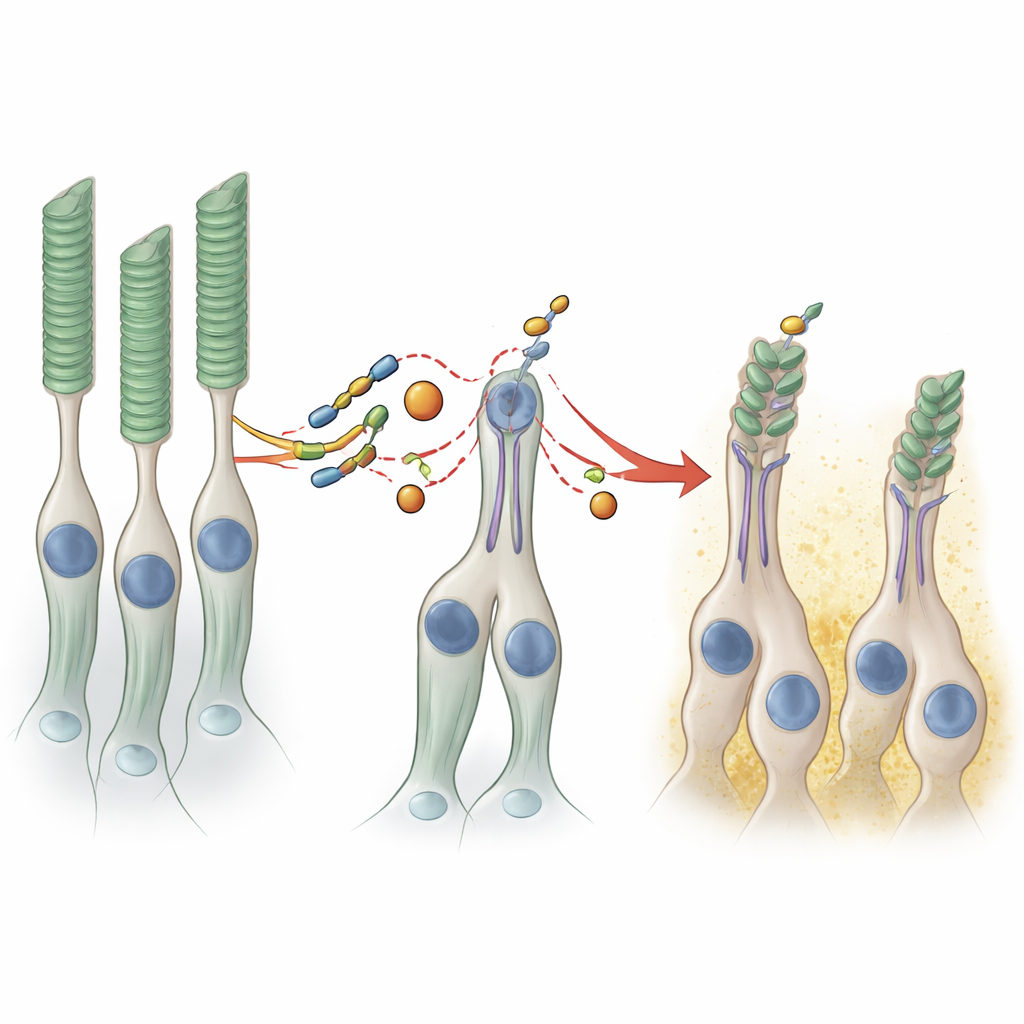
Subtle shifts in RNA processing, big effects on structure
PRPF8 helps run the spliceosome, the cellular machine that edits freshly made RNA messages. The researchers sequenced RNA from the organoids to see how the mutation disrupts this process. Surprisingly, the overall pattern of gene activity was only modestly changed; just two genes were consistently more active in the mutant tissue, and these were linked to support cells such as Müller glia and bipolar cells. The more telling changes lay in how certain RNAs were spliced. Hundreds of introns—the noncoding segments usually removed—were retained at slightly higher rates in the mutant organoids, especially those with weaker “splice sites.” A handful of exons were skipped or included more often. Notably, one affected gene, IFT122, is part of the transport machinery that shuttles key proteins along the tiny bridge between the inner and outer segments of photoreceptors. Faults in this transport system are already known to cause retinal degeneration, suggesting that even small splicing errors in such genes can undermine the long, delicate outer segments.
Circular RNAs as early warning signals
The team also examined circular RNAs, a stable but still mysterious class of RNA molecules produced when the RNA ends are joined to form a loop. In the mutant organoids, more than a hundred circular RNAs changed in abundance compared to controls, with some increasing as the organoids aged. When the researchers compared these patterns to a mouse model carrying the same mutation, they found overlapping host genes that produced misregulated circular RNAs in both species. Because circular RNAs are long-lived and accumulate over time, their altered levels could serve as sensitive markers of disturbed splicing and early retinal stress before massive cell loss occurs.
What this means for people with retinitis pigmentosa
Together, these findings show that a seemingly mild PRPF8 mutation can quietly weaken photoreceptor outer segments in human-like retinal tissue, even when overall gene expression changes are subtle. The work strengthens the idea that, in at least some patients, the disease begins within the light-sensing cells themselves, driven by precise splicing errors in a limited set of vulnerable genes involved in cilia and protein transport. At the same time, shifts in circular RNA levels emerge as a shared signature of such splicing-related forms of retinitis pigmentosa. In the future, pinpointing the key mis-spliced targets and tracking circular RNA markers could guide new therapies aimed at correcting RNA processing and preserving sight.
Citation: Zimmann, F., Banik, P., Kubovčiak, J. et al. PRPF8-associated retinitis pigmentosa variant induces human neural retina-autonomous photoreceptor defects. Sci Rep 16, 10264 (2026). https://doi.org/10.1038/s41598-026-40376-y
Keywords: retinitis pigmentosa, photoreceptor degeneration, RNA splicing, retinal organoids, circular RNA