Clear Sky Science · de
PRPF8-assoziierte Retinitis-pigmentosa-Variante verursacht autonome Photorezeptor-Defekte im menschlichen neuronalen Netzhautgewebe
Warum das für das Sehvermögen wichtig ist
Retinitis pigmentosa ist eine führende Ursache für vererbte Erblindung, doch bei vielen Betroffenen verstehen wir nicht vollständig, warum ihre lichtempfindlichen Zellen zugrunde gehen. Diese Studie nutzt miniaturisierte, im Labor gezüchtete menschliche Netzhäute, um eine rätselhafte Form der Erkrankung zu untersuchen, die durch eine dezente Veränderung im Gen PRPF8 verursacht wird. Indem die Forschenden den Zustand in einer Schale nachbilden, zeigen sie, dass der Schaden im neuronalen Teil der Netzhaut selbst entstehen kann – besonders in den lichtsensitiven Zellen – und nicht nur durch Stützzellen im hinteren Auge ausgelöst werden muss. Ihre Arbeit offenbart außerdem frühe RNA-Veränderungen, die eines Tages helfen könnten, diese langsam verlaufende Erkrankung zu diagnostizieren oder zu verfolgen.
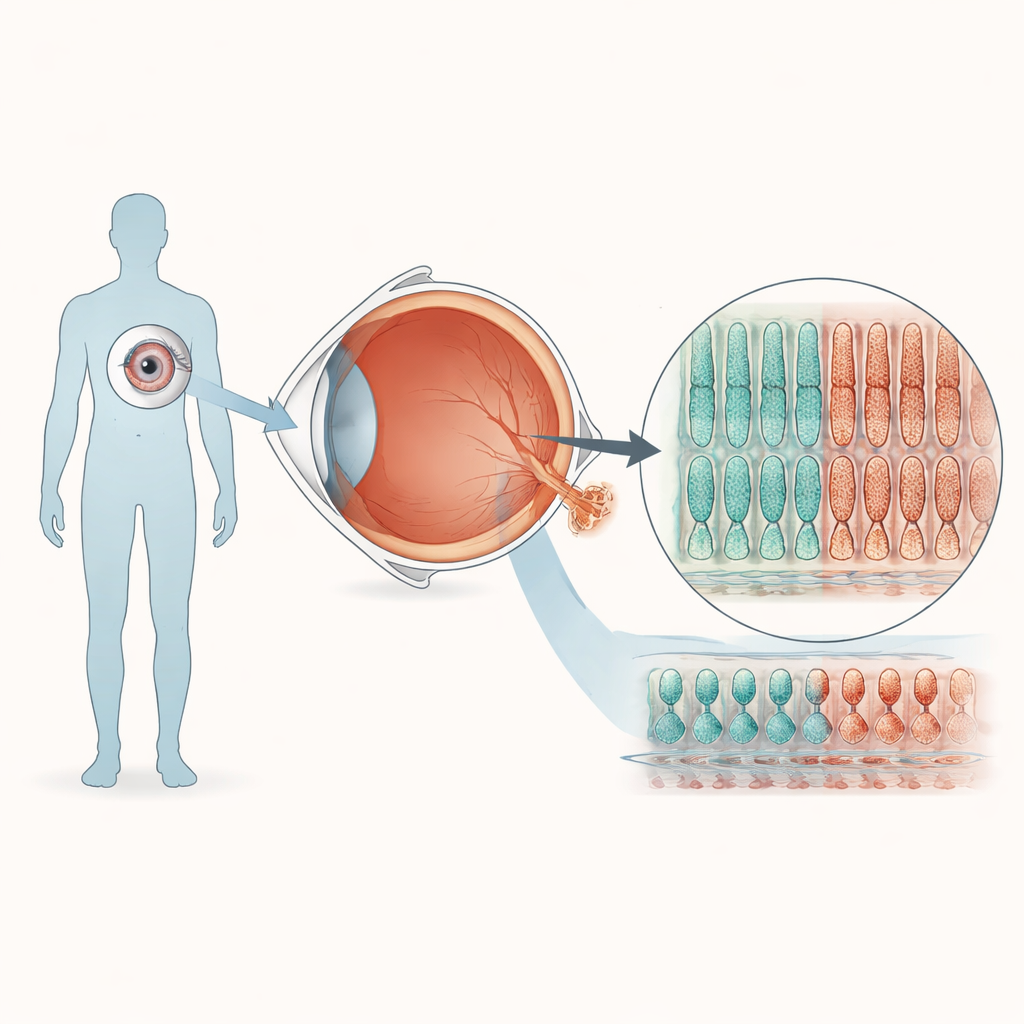
Kleine menschliche Netzhäute im Labor herstellen
Um zu erforschen, wie eine einzelne Basenänderung in PRPF8 das Sehen beeinträchtigt, verwandelte das Team Blutzellen eines gesunden Spenders in induzierte pluripotente Stammzellen, die sich zu vielen Geweben differenzieren lassen. Mit CRISPR-Geneditierung erzeugten sie eine Zelllinie mit der in Retinitis pigmentosa beobachteten PRPF8-Mutation und eine passende Kontrolllinie ohne diese Veränderung. Beide Linien wurden dann über ein monatelanges Protokoll dazu gebracht, sich zu dreidimensionalen retinalen „Organoiden“ selbst zu organisieren, die viele Eigenschaften der menschlichen Netzhaut nachahmen. Unter dem Mikroskop entwickelten Organoide aus mutanten und Kontrollzellen ähnliche geschichtete Strukturen und enthielten die wichtigsten Netzhautzelltypen, was darauf hindeutet, dass die frühe Augenentwicklung trotz der Mutation weitgehend normal verläuft.
Versteckte Schwäche in den lichtempfindlichen Zellen
Bei genauerer Untersuchung zeigte sich jedoch, dass Organoide mit der PRPF8-Mutation weniger von mehreren Proteinen produzierten, die charakteristisch für Photorezeptoren sind – die Zellen, die Licht erfassen. Nachdem die Organoide länger als acht Monate ausreifen durften, zeigten Elektronen- und Lichtmikroskopie einen auffälligen strukturellen Unterschied. In den Kontrollorganoiden war die Oberfläche dicht mit inneren und äußeren Segment-ähnlichen Fortsätzen bedeckt, den spezialisierten Kompartimenten, in denen Photorezeptoren ihre visuellen Pigmente konzentrieren. In den mutanten Organoiden waren diese Strukturen spärlicher, und eine bildbasierte Analyse mittels maschinellen Lernens ergab, dass die „Bürstensaum“-Struktur aus Photorezeptorsegmenten etwa halb so dick war. Da dieses System nahezu kein retinales Pigmentepithel enthält und die beiden Gewebe physisch getrennt sind, deuten diese Defekte auf eine intrinsische Verwundbarkeit der Photorezeptoren selbst bei dieser Form der Erkrankung hin.
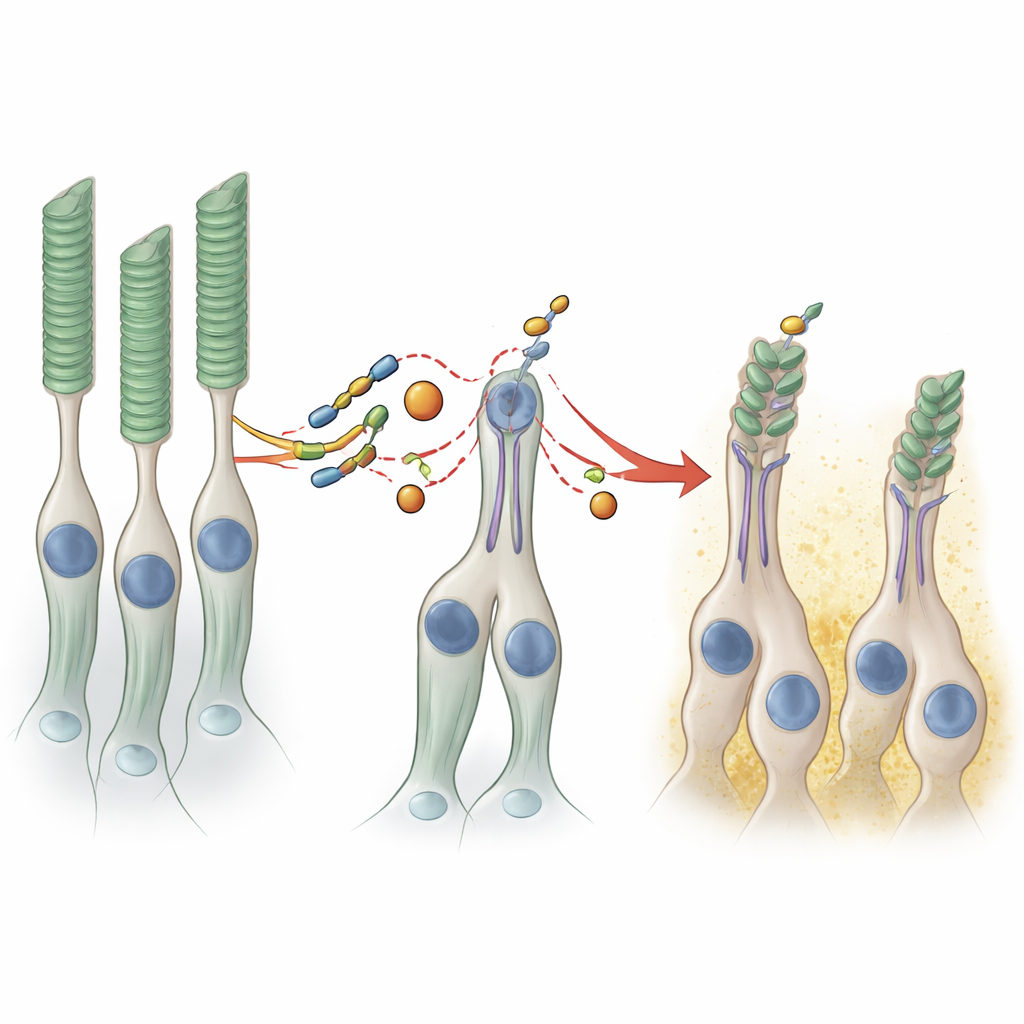
Feine Verschiebungen in der RNA-Verarbeitung, große Auswirkungen auf die Struktur
PRPF8 unterstützt den Ablauf des Spleißosoms, der zellulären Maschinerie, die frisch gebildete RNA-Botschaften schneidet und zusammenfügt. Die Forschenden sequenzierten die RNA aus den Organoiden, um zu sehen, wie die Mutation diesen Prozess stört. Überraschenderweise war das Gesamtmuster der Genaktivität nur mäßig verändert; nur zwei Gene zeigten in dem mutanten Gewebe durchgehend eine höhere Aktivität, und diese standen im Zusammenhang mit Stützzellen wie Müller-Glia und bipolaren Zellen. Aussagekräftiger waren die Änderungen in der Art, wie bestimmte RNAs gespleißt wurden. Hunderte von Introns – die nicht-kodierenden Abschnitte, die normalerweise entfernt werden – wurden in den mutanten Organoiden etwas häufiger belassen, vor allem solche mit schwächeren „Spleißstellen“. Eine Handvoll Exons wurde häufiger übersprungen oder häufiger eingebaut. Bemerkenswerterweise ist eines der betroffenen Gene, IFT122, Teil der Transportmaschinerie, die wichtige Proteine entlang der winzigen Brücke zwischen inneren und äußeren Segmenten der Photorezeptoren befördert. Fehler in diesem Transportsystem sind bereits als Ursache retinaler Degeneration bekannt, was nahelegt, dass schon kleine Spleißfehler in solchen Genen die langen, empfindlichen äußeren Segmente untergraben können.
Zirkuläre RNAs als Frühwarnsignale
Das Team untersuchte auch zirkuläre RNAs, eine stabile, aber noch rätselhafte Klasse von RNA-Molekülen, die entstehen, wenn die RNA-Enden zu einer Schleife verbunden werden. In den mutanten Organoiden veränderten sich mehr als hundert zirkuläre RNAs in der Häufigkeit gegenüber Kontrollen, einige davon nahmen mit dem Altern der Organoide zu. Beim Vergleich dieser Muster mit einem Mausmodell, das dieselbe Mutation trägt, fanden sie überlappende Wirtsgene, die in beiden Arten fehlregulierte zirkuläre RNAs produzierten. Da zirkuläre RNAs langlebig sind und sich im Laufe der Zeit ansammeln, könnten ihre veränderten Mengen als empfindliche Marker gestörter Spleißvorgänge und frühen retinalen Stresses dienen, noch bevor massiv Zellen verloren gehen.
Was das für Menschen mit Retinitis pigmentosa bedeutet
Zusammen zeigen diese Befunde, dass eine scheinbar milde PRPF8-Mutation die äußeren Photorezeptorsegmente in menschenähnlichem Netzhautgewebe stillschweigend schwächen kann, selbst wenn die Veränderungen in der Genexpression insgesamt subtil sind. Die Arbeit untermauert die Idee, dass die Erkrankung bei zumindest einigen Patienten innerhalb der lichtempfindlichen Zellen selbst beginnt, getrieben von präzisen Spleißfehlern in einer begrenzten Menge besonders anfälliger Gene, die an Zilien und Proteintransport beteiligt sind. Gleichzeitig treten Veränderungen in den zirkulären RNA-Leveln als gemeinsames Merkmal solcher spleißbedingter Formen der Retinitis pigmentosa hervor. Künftig könnten das gezielte Ausmachen der wichtigsten fehlgespleißten Ziele und das Verfolgen zirkulärer RNA-Marker neue Therapien leiten, die auf die Korrektur der RNA-Verarbeitung abzielen und das Sehen erhalten.
Zitation: Zimmann, F., Banik, P., Kubovčiak, J. et al. PRPF8-associated retinitis pigmentosa variant induces human neural retina-autonomous photoreceptor defects. Sci Rep 16, 10264 (2026). https://doi.org/10.1038/s41598-026-40376-y
Schlüsselwörter: Retinitis pigmentosa, Photorezeptor-Degeneration, RNA-Spleißen, Retinale Organoide, zirkuläre RNA