Clear Sky Science · fr
Assemblages de génomes de haute qualité de deux souches de Prototheca wickerhamii
Pourquoi cette minuscule algue compte pour notre santé
La plupart d’entre nous imaginent les algues comme un dépôt vert inoffensif sur les étangs, alimenté par la lumière du soleil. Mais certains apparentés des algues ont perdu leur pigment vert et sont devenus des agents furtifs capables d’infecter les humains et les animaux. L’un de ces coupables, Prototheca wickerhamii, provoque des infections rares mais tenaces de la peau, des tissus mous et parfois d’organes plus profonds. Les médecins ont du mal à la combattre en partie parce que sa biologie fondamentale reste mal comprise. Cette étude fournit des plans d’ADN de haute qualité pour deux souches cliniques de ce microbe, offrant aux chercheurs une liste détaillée des composants qui peut aider à expliquer comment il survit dans l’organisme et comment nous pourrions mieux diagnostiquer et traiter les infections qu’il cause. 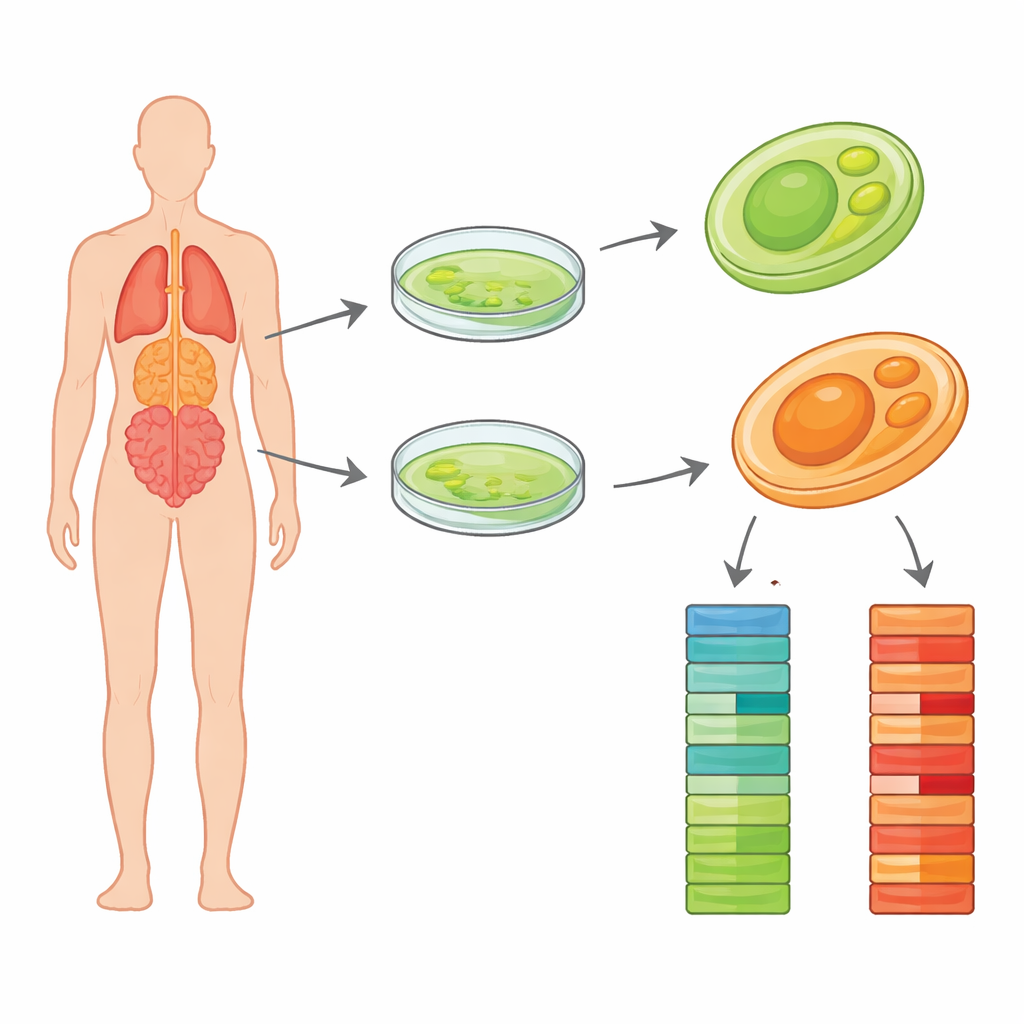
Un cousin incolore caché en pleine vue
Prototheca wickerhamii appartient à un groupe peu connu de microalgues « incolores » qui ne réalisent plus la photosynthèse. Plutôt que de vivre de la lumière comme leurs parents verts, elles se contentent d’environnements humides et parfois d’hôtes à sang chaud. Au cours des deux dernières décennies, les infections signalées causées par ces organismes ont augmenté, en particulier chez les personnes immunodéprimées et chez les animaux de compagnie. Pourtant, le véritable fardeau est probablement sous‑estimé, car Prototheca peut être manquée ou mal identifiée dans les tests de laboratoire courants. Des travaux antérieurs ont décodé l’ADN d’une souche de référence et ont suggéré que l’organisme porte de nombreux gènes similaires à des facteurs de virulence connus chez des champignons pathogènes, laissant penser que son génome s’est adapté à la vie dans le corps humain.
Collecter et lire l’ADN du microbe
Dans la nouvelle étude, les scientifiques se sont concentrés sur deux souches cliniques, nommées Pw26 et PwS1, isolées chez des patients de différentes villes chinoises. Ils ont d’abord cultivé des colonies pures sur des milieux de laboratoire standard et confirmé l’absence d’autres microbes dans les cultures. L’équipe a ensuite extrait de l’ADN de haute qualité et utilisé une méthode moderne de lectures longues appelée séquençage PacBio HiFi. Contrairement aux techniques plus anciennes qui fragmentent l’ADN en très courts morceaux, les lectures HiFi couvrent des dizaines de milliers de bases à la fois avec une grande précision. Cela facilite la reconstruction de chromosomes entiers avec peu de lacunes. Les chercheurs ont généré plus d’un milliard et demi de bases de séquence pour Pw26 et plus de huit cents millions pour PwS1, fournissant une couverture profonde pour les deux génomes.
Assembler des génomes complets et repérer les motifs répétés
À l’aide de logiciels d’assemblage spécialisés, les longues lectures d’ADN ont été cousues en segments continus représentant les chromosomes de l’organisme. Les tailles finales des génomes étaient d’environ 17,8 millions et 17,4 millions de bases pour Pw26 et PwS1 — similaires, mais légèrement supérieures, à la souche étudiée précédemment. Chacun a été assemblé en seulement 14 à 17 fragments, et des contrôles statistiques ont montré que la plupart des gènes centraux attendus étaient présents, signe de complétude. L’équipe a ensuite recherché des éléments d’ADN répétés, qui peuvent façonner l’évolution des génomes. Ces répétitions représentaient environ 6 % de Pw26 et 4 % de PwS1, dominées par une classe appelée long terminal repeat (LTR) souvent observée dans les génomes de plantes et d’algues. De subtiles différences dans la quantité et le type de répétitions entre les deux souches peuvent refléter la façon dont chacune s’est adaptée à des environnements ou à des hôtes différents.
Ce que les gènes révèlent sur le mode de vie du microbe
Après avoir masqué les répétitions, les chercheurs ont prédit les gènes codant des protéines en combinant trois approches : des modèles informatiques entraînés à reconnaître la structure des gènes, la comparaison avec des protéines connues d’algues apparentées et d’autres souches de Prototheca, et l’alignement de données d’ARN précédemment collectées. Cela a permis d’identifier environ 6 400 gènes dans chaque génome. Ils ont ensuite annoté ces gènes à l’aide de deux catalogues largement utilisés de fonctions géniques. Le premier, appelé Gene Ontology, classe les gènes selon les types de tâches qu’ils accomplissent dans la cellule, tandis que la base KEGG les relie aux voies métaboliques. Les deux souches possédaient de nombreux gènes impliqués dans la production d’énergie, la dégradation et la synthèse des nutriments, et la régulation des processus cellulaires. PwS1 montrait une mise en avant des voies liées aux lipides et à la signalisation, faisant écho à des observations antérieures qui associaient l’aspect mucoïde inhabituel de cette souche et sa moindre toxicité à des modifications de sa surface et de son métabolisme. 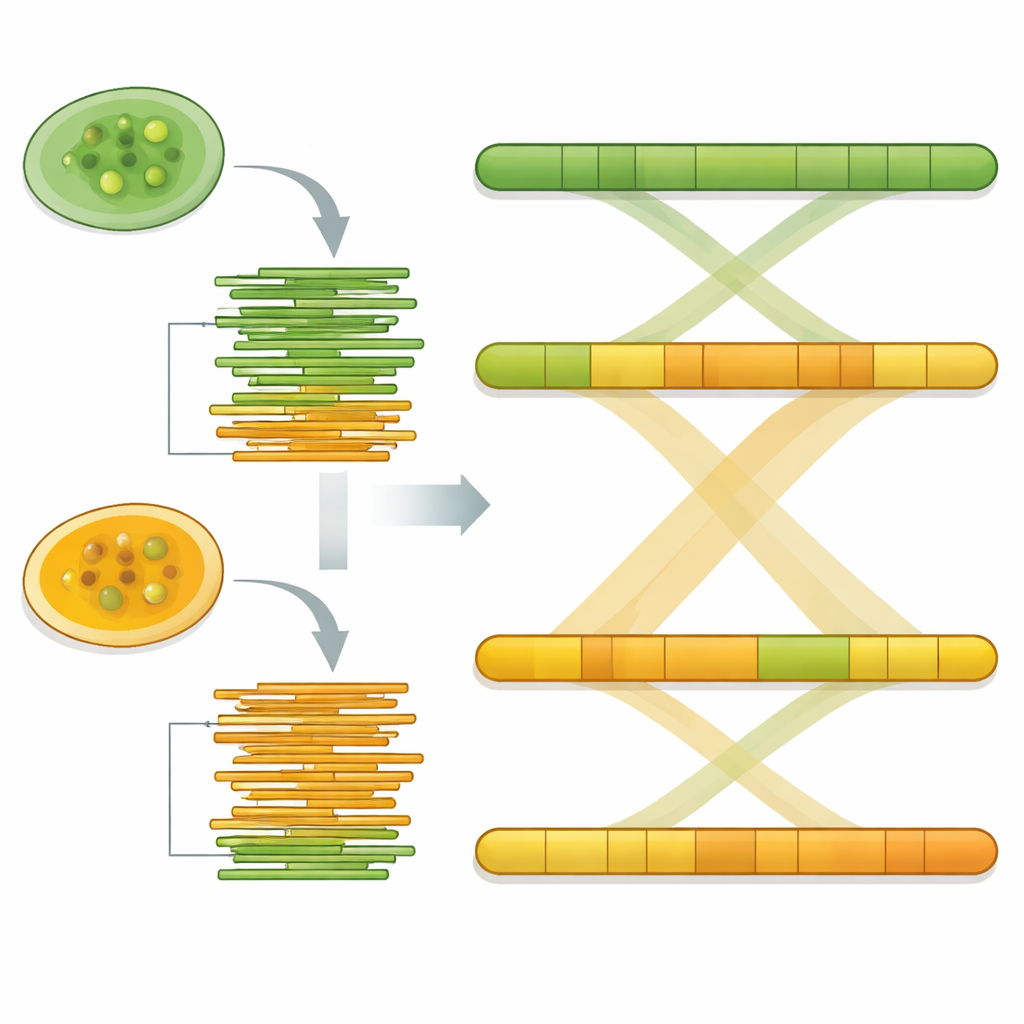
Vérifier la précision et comparer les deux souches
Pour s’assurer que leurs reconstructions étaient fiables, l’équipe a réaligné les lectures longues originales sur chaque génome assemblé. Plus de 93 % des lectures se sont remappées avec une couverture homogène, et la composition de bases n’a montré aucun signe de contamination. Un autre contrôle de qualité, appelé BUSCO, a confirmé que plus de 86 % d’un jeu standard de gènes algaux conservés étaient présents et intacts dans les deux souches. Enfin, lorsque les deux génomes ont été comparés à l’échelle entière à l’aide d’outils de comparaison, leurs segments d’ADN correspondaient presque un pour un, indiquant un très haut degré de similarité et soutenant l’idée que les assemblages reflètent fidèlement les chromosomes sous-jacents.
Ce que cela signifie pour le diagnostic et le traitement à venir
Pour les non‑spécialistes, le message principal est que nous disposons désormais de cartes d’ADN détaillées et fiables pour deux souches pathogènes de Prototheca wickerhamii. Ces cartes ne guérissent pas les infections en elles‑mêmes, mais elles fournissent la base pour poser des questions plus précises : quels gènes permettent au microbe d’échapper au système immunitaire, quelles voies pourraient être ciblées par des médicaments existants, et comment les différentes souches varient en virulence et en réponse aux traitements ? Comme les données ont été rendues publiques, des laboratoires du monde entier peuvent les utiliser pour concevoir de meilleurs tests diagnostiques, suivre les éclosions dans une approche One Health reliant santé humaine et animale, et in fine guider des stratégies thérapeutiques plus ciblées pour cet agent peu fréquent mais difficile à traiter.
Citation: Fang, L., Guo, J., Ning, Q. et al. High-Quality Genome Assemblies of Two Prototheca wickerhamii Strains. Sci Data 13, 633 (2026). https://doi.org/10.1038/s41597-026-06916-x
Mots-clés: Prototheca wickerhamii, assemblage de génome, infection opportuniste, séquençage long-courrier, génomique des agents pathogènes