Clear Sky Science · fr
Une approche par la théorie de l'information pour quantifier la réponse dépendante de la séquence des moteurs d'acides nucléiques avec des applications au séquençage de l'ADN par nanopore
Lire l'ADN avec de toutes petites machines
Toutes les cellules de votre corps dépendent de machines microscopiques qui se déplacent le long de l'ADN pour le copier et le réparer. Les mêmes types de machines alimentent aujourd'hui des dispositifs de séquençage de l'ADN rapides. Cette étude pose une question apparemment simple mais aux grandes implications : au‑delà des signaux électriques habituellement exploités en séquençage par nanopore, quelle quantité d'information supplémentaire sur la séquence d'ADN est cachée dans la manière dont ces minuscules moteurs se déplacent — combien de temps ils font des pauses et à quelle fréquence ils reculent ? En utilisant des outils de la théorie de l'information, les auteurs montrent que ces mouvements subtils peuvent augmenter de façon substantielle la précision de lecture de l'ADN, et décrivent comment concevoir de meilleurs moteurs moléculaires pour les technologies de séquençage futures.
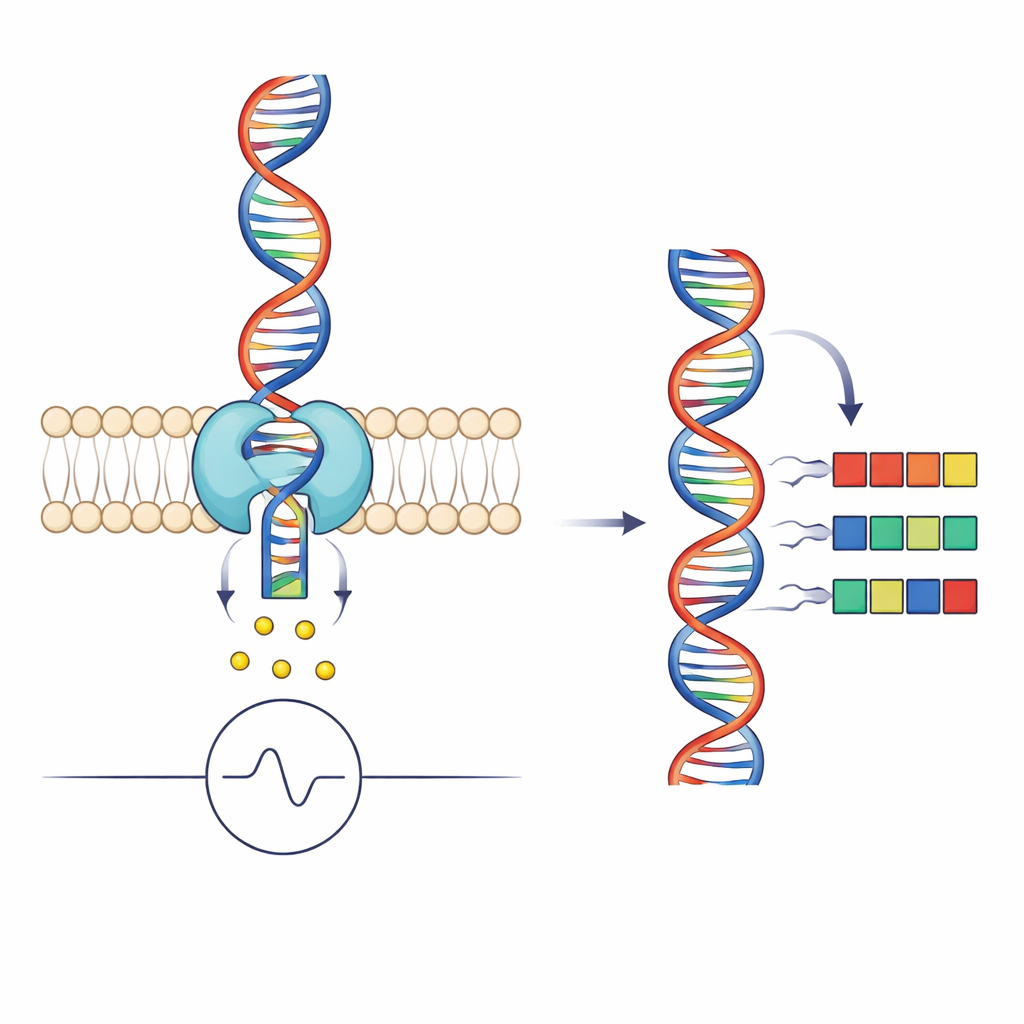
Comment les nanopores transforment l'ADN en signaux
Le séquençage par nanopore consiste à faire passer un brin d'ADN unique à travers un minuscule trou dans une membrane tout en mesurant le flux d'ions à travers le pore. Lorsque différents groupes de bases occupent la partie la plus étroite du pore, ils bloquent partiellement le courant de manières distinctes, produisant un motif électrique caractéristique que l'on peut décoder en séquence. Une enzyme motrice, ici une hélicase appelée Hel308, saisit l'ADN et l'alimente par petits pas à travers le pore. Chaque « lecture » de séquençage n'est donc pas seulement une trace de courant mais aussi un enregistrement détaillé du mouvement de l'hélicase : combien de temps elle attend à chaque position et si elle recule occasionnellement.
Utiliser la théorie de l'information pour mesurer des indices cachés
Les auteurs utilisent le concept d'information mutuelle pour quantifier dans quelle mesure la séquence d'ADN contrôle différents observables : le courant ionique, le temps que Hel308 passe à chaque pas, et sa probabilité de reculer. L'information mutuelle, mesurée en bits, répond à la question : en moyenne, combien peut‑on apprendre sur une base d'ADN à partir d'un signal donné ? En analysant des milliers de mesures, ils trouvent que le courant ionique est le plus sensible à un court segment d'environ quatre bases situé dans l'étranglement du pore, tandis que le mouvement de Hel308 est principalement gouverné par des bases situées à 16–21 positions en amont. En particulier, des bases à deux positions (environ 17 et 20 nucléotides depuis le pore) influencent fortement la durée de séjour de l'enzyme et la probabilité qu'elle recule. La combinaison du temps de séjour et du comportement de recul révèle plus d'information sur ces bases que chacune des caractéristiques prise séparément.
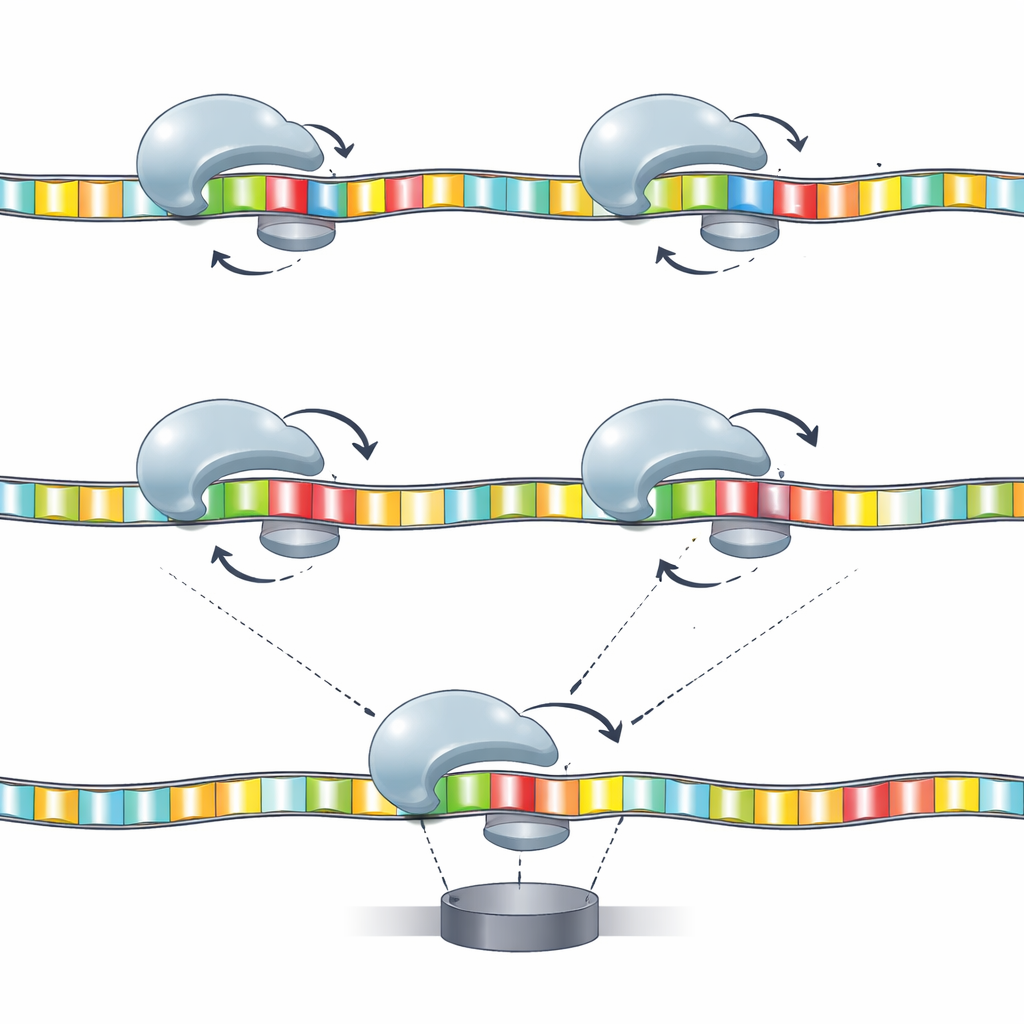
Construire des cartes du mouvement vers la séquence
Les appareils de séquençage reposent souvent sur des modèles de « k‑mer », qui lient un petit groupe de k bases voisines à un signal caractéristique. Ici, les auteurs adaptent cette idée au mouvement de l'hélicase. Ils construisent des modèles dans lesquels des paires ou triplets spécifiques de bases à des positions clés déterminent conjointement le profil des temps de séjour et des reculs. La théorie de l'information montre que certaines combinaisons — comme les bases aux positions 17 et 20, ou des trimères incluant les positions 16, 17 et 20 — portent beaucoup plus d'information que n'importe quelle base prise isolément. Autrement dit, l'enzyme ne « ressent » pas une seule base à la fois ; elle répond à de petits motifs de séquence répartis le long du brin, et ces réponses peuvent être cartographiées systématiquement.
Des simulations de séquençage montrent un gain important
Pour tester l'utilité pratique de cette information basée sur le mouvement, l'équipe a simulé des runs de séquençage par nanopore en utilisant des modèles réalistes du courant et de la cinétique. Ils ont ensuite utilisé un algorithme de décodage pour reconstruire des séquences d'ADN à partir de trois types d'entrée : courant seul, cinétique seule, ou les deux combinés. Le courant ionique seul donne déjà de bonnes performances, tandis que la cinétique seule est moins précise. Mais lorsque les deux sont combinés, les taux d'erreur chutent fortement — d'environ quatre à cinq fois en moins à haute couverture comparé à l'utilisation du courant seul. Notamment, un nombre modeste de lectures exploitant les deux signaux peut surpasser de nombreuses lectures ne tenant compte que du courant, ce qui suggère un séquençage plus rapide et plus précis si les données cinétiques sont pleinement exploitées.
Ajuster le moteur moléculaire lui‑même
Les chercheurs examinent également comment modifier l'hélicase pourrait améliorer encore les performances. Guidés par des données structurelles, ils ont muté des acides aminés individuels de Hel308 en contact avec l'ADN et observé comment ces changements affectaient les temps de séjour et les reculs. La plupart des mutations ont eu peu d'effet, mais quelques‑unes ont provoqué de larges décalages systématiques dans la durée de l'enzyme et la fréquence des reculs, tout en préservant sa sensibilité à la séquence. Deux positions de la protéine, en particulier, reflétaient les positions de séquence clés mises en évidence par l'analyse d'information, suggérant un lien direct entre des acides aminés spécifiques et le comportement de détection de séquence de l'enzyme. L'étude montre aussi qu'il existe un compromis : un mutant qui transporte légèrement plus d'information par pas se déplace plus lentement, de sorte que son information totale par seconde est similaire à celle de l'enzyme d'origine.
Pourquoi cela compte pour la lecture future de l'ADN
Pour un non‑spécialiste, l'essentiel est que les séquenceurs par nanopore peuvent faire plus que lire des motifs électriques de l'ADN ; ils peuvent aussi « écouter » comment le moteur moléculaire se comporte en marchant le long du brin. Ce travail fournit une méthode rigoureuse pour mesurer la quantité d'information de séquence supplémentaire portée par ce mouvement et montre que l'inclure pourrait grandement améliorer la précision, en particulier dans des régions difficiles comme les répétitions ou pour des alphabets génétiques étendus. En utilisant la théorie de l'information comme outil de conception et de criblage, les scientifiques peuvent ingénier systématiquement des enzymes motrices dont les pauses et hésitations rendent les séquences d'ADN plus faciles à lire, ouvrant la voie à des technologies de séquençage plus rapides, plus fiables et plus polyvalentes.
Citation: Craig, J.M., Laszlo, A.H., Brinkerhoff, H. et al. An information theory approach to quantifying the sequence-dependent response of nucleic acid motors with applications to nanopore DNA sequencing. Nat Commun 17, 3231 (2026). https://doi.org/10.1038/s41467-026-69867-2
Mots-clés: séquençage par nanopore, cinétique des hélicases, théorie de l'information, enzymes motrices de l'ADN, modèles k-mer