Clear Sky Science · en
Computational identification of conserved dengue virus envelope protein epitopes for vaccine design and immunodiagnostic platform
Why this research matters
Dengue fever is a mosquito-borne disease that now threatens nearly half the world’s population. Yet doctors still lack a vaccine that safely protects against all four versions of the virus and blood tests that can reliably tell them which version is causing an infection. This study uses advanced computer tools to scan the virus in detail, hunting for tiny pieces that could form the basis of a safer, broader vaccine and more precise diagnostic tests.
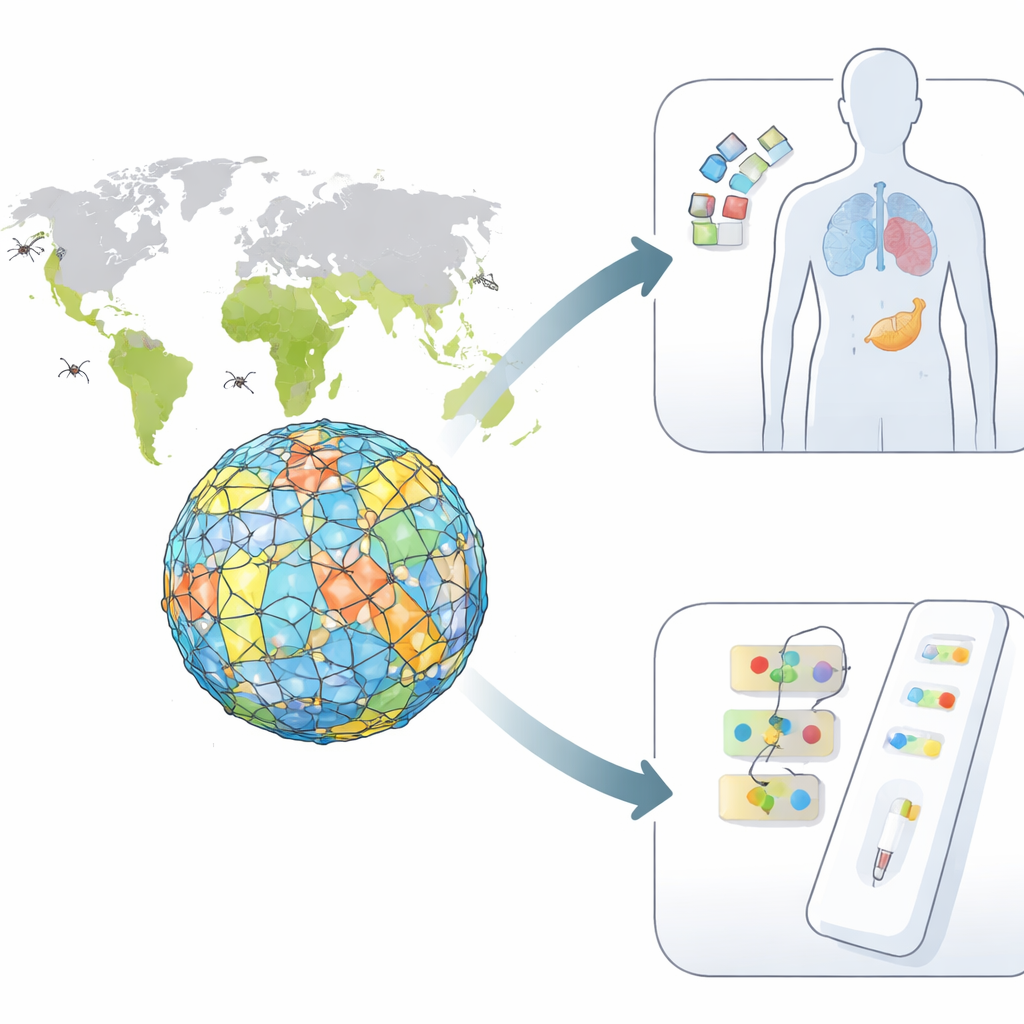
Four similar viruses, one big problem
The dengue virus comes in four closely related forms, or serotypes, each capable of causing anything from mild fever to life-threatening illness. Infection with one serotype protects you only against that one and can even worsen disease if you are later infected with a different type. Existing vaccines struggle because they must trigger strong, balanced protection against all four at once, without tipping the immune system into harmful overreaction. On top of this, current blood tests can confuse dengue with related viruses such as Zika, making it hard to track outbreaks accurately.
Zooming in on the virus coat
The researchers focused on the virus’s envelope protein, a major component of the outer coat that the immune system “sees” first. They collected thousands of envelope protein sequences from all four serotypes and aligned them to find regions that are either highly conserved (similar across all types) or clearly different from one type to another. The goal was twofold: conserved regions might serve as the backbone of a universal vaccine, while unique regions could act as fingerprints for tests that distinguish between serotypes.
Finding promising targets with computers
Using a suite of immunoinformatics tools, the team predicted which short stretches of the envelope protein are likely to be recognized by the body’s T cells and B cells—the white blood cells that coordinate and execute immune attacks. They evaluated each candidate segment for how strongly it might trigger an immune response, whether it resembles any human proteins (which could cause autoimmunity), and whether it is likely to be toxic or allergenic. Several segments stood out as both safe and strongly visible to the immune system, including a set shared across all serotypes that could underpin a “tetravalent” dengue vaccine and other sets that are unique to dengue types 2, 3, or 4, ideal for highly specific diagnostic tests.
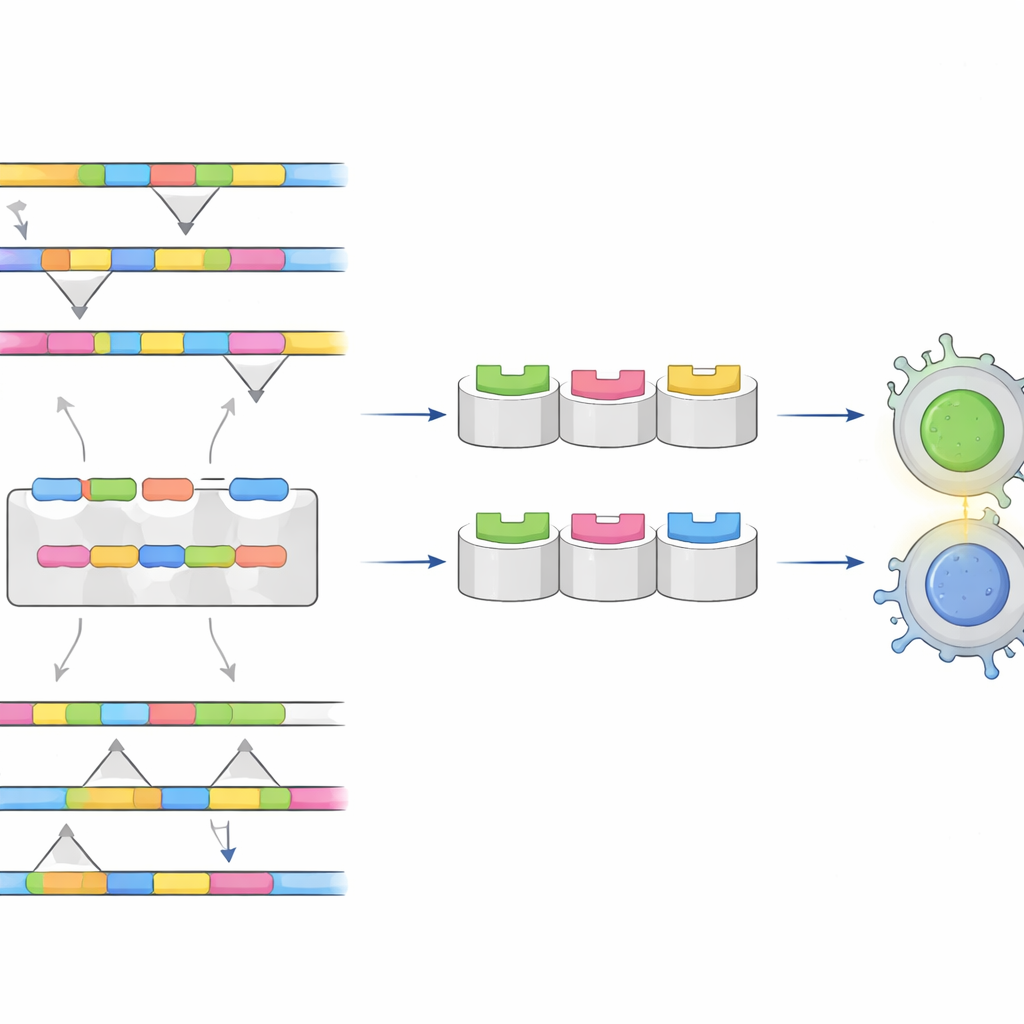
Testing the fit in 3D
To go beyond sequence patterns, the researchers built three-dimensional models of how these viral segments would slot into human immune molecules that present them to T cells. They used molecular docking to simulate the physical fit between peptide and immune receptor, and normal mode analysis to probe how stable these complexes would be as they flex and move. Some peptide–receptor pairs formed especially tight, stable bindings, suggesting they could reliably alert T cells across many people worldwide. A population analysis, based on how common different immune receptor variants are in various regions, indicated that the best set of candidates could, in theory, cover about two-thirds of the global population, with particularly strong coverage in East Asia, Europe, and the Americas.
What this means for future tools
In plain terms, this work delivers a carefully filtered shortlist of viral “pieces” that look especially promising for next-generation dengue vaccines and blood tests. The conserved segments could help build a single vaccine that works across all four dengue types, while the unique segments could allow doctors to tell exactly which type is causing a patient’s illness. Although these predictions still need to be tested in the lab and in clinical studies, the study shows how computer-driven analysis can greatly speed and sharpen the search for vaccine and diagnostic targets against a rapidly spreading tropical disease.
Citation: da Silva, M.K., Fulco, U.L., Alqahtani, T. et al. Computational identification of conserved dengue virus envelope protein epitopes for vaccine design and immunodiagnostic platform. Sci Rep 16, 14167 (2026). https://doi.org/10.1038/s41598-026-37744-z
Keywords: dengue vaccine, epitope mapping, envelope protein, immunoinformatics, serotype-specific diagnostics