Clear Sky Science · zh
用于犬类摩擦病毒全基因组恢复和分子流行病学的多重NGS面板
这对宠物和人类为何重要
爱犬人士、野生动物爱好者以及公共卫生观察者都与一种名为犬瘟热的病毒息息相关。这种高度传染性的疾病能对宠物犬和野生食肉动物造成毁灭性打击,其近亲还包括人类的麻疹。这里描述的研究推出了一种新的、经济的基因检测方法,能直接从患病动物中读取犬瘟病毒的全部遗传密码,帮助科学家追踪其传播、变异,以及未来可能威胁到新物种的风险。
跨越物种边界的病毒
犬瘟热病毒是一个全球性的威胁,能感染家犬、狐狸和大型猫科等野生食肉动物,甚至一些非食肉物种。它攻击多个器官,导致发热、呼吸问题、肠道疾病,有时还会造成脑部损伤。尽管它目前尚不是人类疾病,但实验显示该病毒能够适应并使用与人类麻疹相同的细胞“通道”。这使得监测该病毒在不同宿主和地区的演化变得重要,既为保护动物,也为预判那些罕见但严重的行为转变做准备。
从片段快照到完整基因画像
到目前为止,大多数关于该病毒的研究只读取其一个基因,称为H基因,该基因帮助病毒附着细胞。观察这段单一序列有助于将病毒分组为谱系并追踪大致的传播模式。但它忽略了超过15,000个控制病毒复制、逃避免疫系统及适应新宿主的遗传“字母”。完整基因组信息远为丰富,但从常规临床样本中获取却很困难,尤其是在资源有限的环境以及如拉丁美洲多数地区这类数据代表性不足的区域。

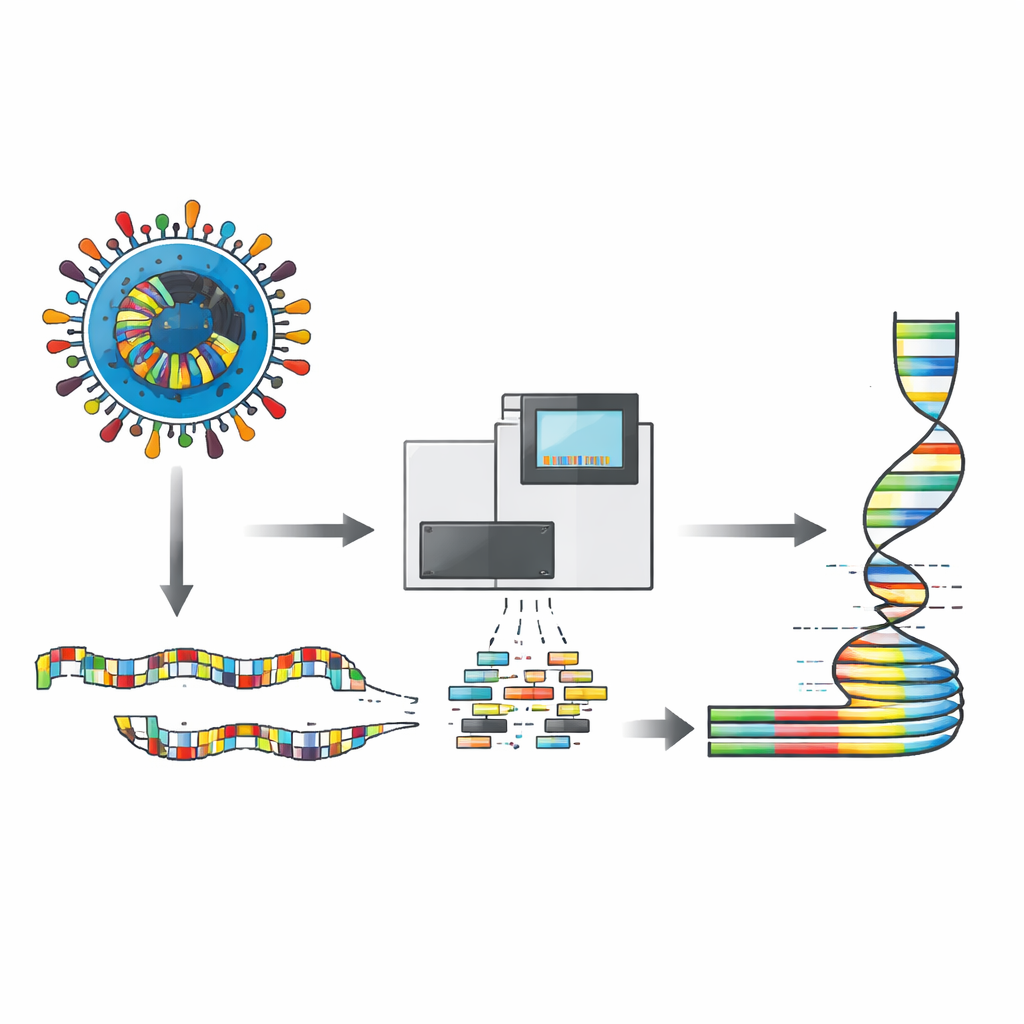
一种用于读取完整病毒基因组的新工具包
研究者开发了一个实验室面板,将病毒基因组分解为许多小而重叠的片段,这些片段可以在同一反应中同时扩增,然后由Illumina下一代测序仪读取。设计使用了236个短引物,分为两组,选择时考虑了来自世界各地的犬瘟病毒,使得即便是变异株也能被覆盖。该方案偏好短片段,这些片段更容易从受损或低质量样本中恢复——正是经常从患病或死亡动物采集到的样本类型。在疫苗株及来自玻利维亚、厄瓜多尔、墨西哥、秘鲁和乌拉圭的15只感染犬上测试时,该方法常规覆盖率超过97%,并且在每个位点常常有数千条读数,即便原始样本中病毒量很低。
新基因图谱揭示的发现
有了这些完整序列,团队将15个拉丁美洲病毒与此前可得的173个完整基因组进行了比较。更广的视野使他们能够将每个病毒定位到已知谱系中,并观察到单一基因无法解析的更细致分支模式。来自玻利维亚的犬携带的病毒属于此前在乌拉圭、巴西、阿根廷和智利见到的一个谱系,扩展了该谱系已知的分布范围。墨西哥犬携带的是北美谱系。厄瓜多尔和秘鲁的株则形成了一个独立簇,接近但有别于另一个北美群体,提示区域性分化,随着更多数据的积累可能值得赋予正式名称。当仅使用H基因重复分析时,许多大范围的分组仍成立,但部分关系变得模糊,凸显了全基因组图景的更高分辨力。
单个动物体内的隐匿变异
由于该方法产生非常深的覆盖深度,它还能检测出在单一宿主体内循环但未成为优势的少数病毒变体。研究者在大多数犬样本中发现了这样的少数变体。许多是单个“字母”变更,有些改变了与宿主防御相互作用的病毒蛋白的氨基酸。在墨西哥样本中,表面蛋白上出现了小的缺失,可能改变病毒与细胞结合的方式,从而影响免疫系统的识别。一个乌拉圭样本在参与复制和免疫逃逸的蛋白中带有插入。这些改变有些可能反映了宿主体内真正的病毒“试验”,也有些可能是扩增过程中的伪影。无论哪种情况,这项工作都说明单只犬体内的病毒群体并非单一,而是由略有不同的基因组云构成,这些变体可以为进化提供原料。
未来的意义
对非专业读者而言,关键结论是:科学家现在拥有一种实用且相对低成本的方法,能够直接从野外患病动物读取犬瘟热病毒几乎完整的遗传密码,无需缓慢且专业的细胞培养步骤。这为在数据稀缺的地区开展常规基因组监测打开了大门,有助于我们更好地观察病毒如何在国家之间移动、在宠物与野生动物之间溢出并探索新的遗传可能性。将全基因组追踪与对每个宿主内隐匿变体的观察相结合,这一方法将加强保护犬类、维护脆弱野生动物种群并监控与重大人类疾病同属一科的病毒的努力。
引用: Panzera, Y., Condon, E., Escardó, J. et al. Multiplex NGS Panel for whole-genome recovery and molecular epidemiology of canine morbillivirus. npj Vet. Sci. 1, 5 (2026). https://doi.org/10.1038/s44433-026-00007-8
关键词: 犬瘟热病毒, 病毒基因组学, 多重测序, 野生动物疾病, 一次性健康(One Health)