Clear Sky Science · zh
全基因组精细定位提高了致病变异的识别
为何找到真实的遗传信号很重要
许多常见性状——从身高和体重到精神分裂症和克罗恩病——都受到散布在基因组中成千上万微小 DNA 变异的影响。现代研究能够发现与性状相关的 DNA 区域,但常常无法确定这些区域中哪些具体变异是真正导致效应的因素。本文介绍了一种在全基因组范围内同时扫描以锁定最可能致病变异的新方法,帮助科学家把粗略的“关注社区”转化为 DNA 中更精确的“门牌号”。
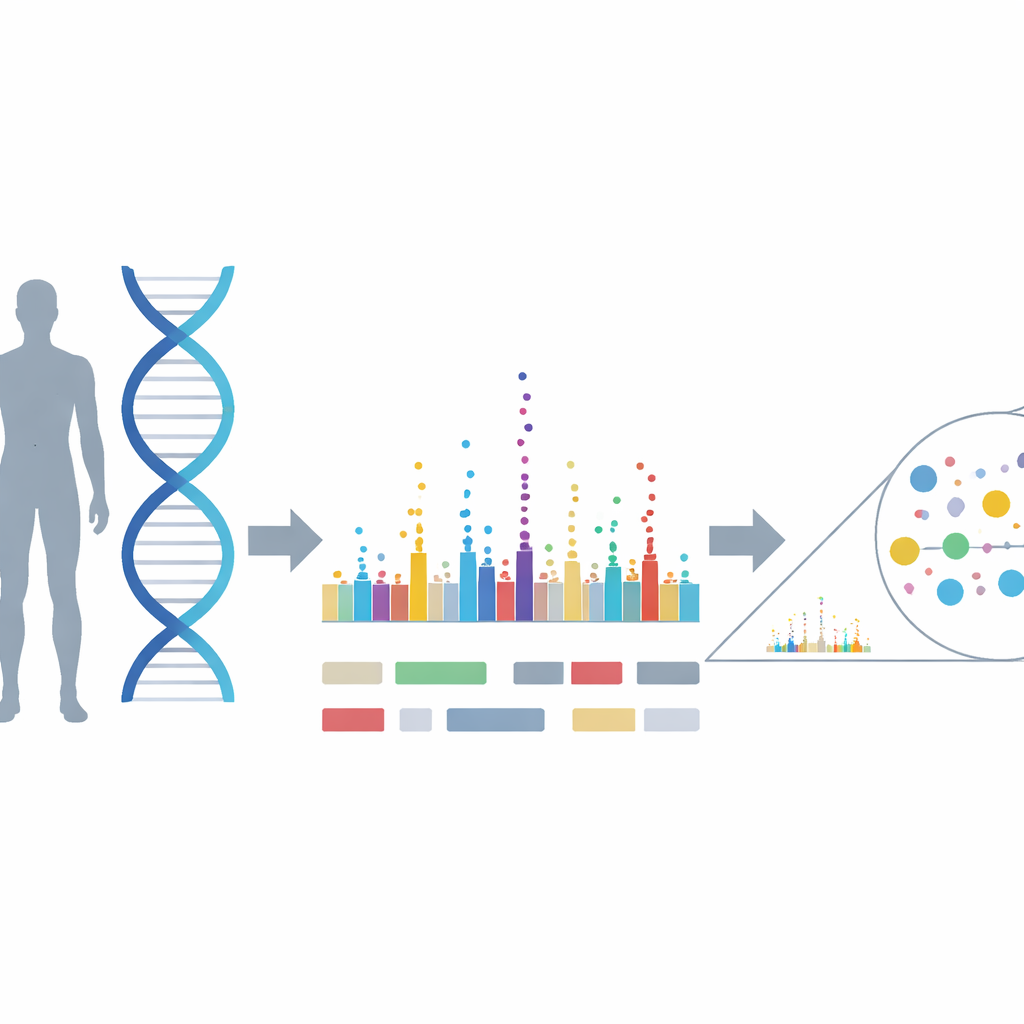
从粗略地图到精确位置
标准的全基因组关联研究(GWAS)在数百万个 DNA 标记与性状之间寻找统计学关联,产生类似天际线的染色体峰图。每个峰都标示了一个社区,其中若干邻近标记由于共同遗传而都显示出与性状相关的迹象。这使得难以判断哪个标记——或哪些标记组合——是真正负责的。传统的精细定位方法通常逐一区域放大分析,只聚焦最高的峰并分别处理这些窗口。这种策略会遗漏许多真实但较弱的信号,在复杂连锁结构的区域表现不佳,并且难以为未来研究需要多大样本量提供指导。
一种用于精确定位变异的全基因组方法
作者提出了一种“全基因组精细定位”方法,对全基因组范围内的所有常见 DNA 标记同时进行分析。他们的关键工具 SBayesRC 使用贝叶斯混合模型:它不再假设每个标记要么重要要么不重要,而是允许标记属于多个效应大小类别,从零到较大不等。更关键的是,方法还借用功能基因组数据的信息——例如某个标记是否位于基因、调控区或进化保守序列中——以便将概率倾向于生物学上更有可能的候选变异。通过对所有标记联合拟合并从这些注释中学习,该方法能更准确地估计哪些变异可能是致病的以及它们效应的大小。
在模拟和真实性状上的性能测试
通过基于真实人类遗传数据的大规模计算机模拟,研究团队将其全基因组方法与广泛使用的逐区工具进行了比较。他们表明 SBayesRC 为每个标记提供了更为校准的概率,捕获了更大比例的真实致病变异,并且其“可信集”(最有可能包含致病变异的小候选组)所含标记更少。将该方法应用于英国生物样本库以及主要精神病和免疫疾病研究的真实数据时,所鉴定的变异在独立样本中更常被复制,并且在不同祖源群体间也能更准确地预测性状。该方法还系统性地发现了位于传统严格 GWAS 显著性阈值之外的重要变异,揭示了标准分析会忽视的信号。
看我们能捕获多少遗传力
因为 SBayesRC 能估计总体遗传架构——致病变异的数量及其效应分布——它可用于前瞻性预测。作者开发了一个功效计算器,可以预测在给定未来样本量下,我们预计能定位多少致病变异以及这些变异应能解释该性状多少比例的遗传影响(基于 SNP 的遗传度)。据此估计,大约两百万参与者规模的研究通常可以精细定位出解释多数复杂性状常见遗传成分一半以上的变异。他们还展示了某些性状(例如血细胞计数)比高度多基因性状(如认知)更易被精细定位,后者可能需要更大的样本量。
真实世界中的致病变异例子
作者突出展示了具体的 DNA 变异以说明该方法的价值。在与肥胖相关的著名 FTO 区域,全基因组精细定位方法正确地优先考虑了先前在实验中被证实影响脂肪生物学的一个变异,这得益于跨物种的保守性信号。在精神分裂症中,该方法提升了那些罕见但功能上更有说服力的变异的优先级,这些变异位于参与脑细胞结构和信号传导的基因中,包括在 ACTR1B 和 SLC39A8 中有强烈蛋白质和细胞类型数据支持的变异。在克罗恩病中,它发现了额外的可能致病变异,这些变异位于经典 GWAS 门槛之下,但在考虑周围标记的联合信息后具有生物学合理性。
这对未来遗传学研究意味着什么
总体而言,研究表明,一次性分析全基因组并整合功能线索,可以更清晰地识别出哪些 DNA 变异对复杂性状真正重要。与其将 GWAS 视为一长串粗略区域,这种方法将其转化为高分辨率的地图,揭示哪些变异值得在实验和药物开发中进行后续研究。通过预测随着样本量增长还能学到多少,该工作为设计未来遗传研究提供了一张路线图,推动我们更接近解释并最终干预常见疾病背后的生物学机制。
引用: Wu, Y., Zheng, Z., Thibaut, L. et al. Genome-wide fine-mapping improves identification of causal variants. Nat Genet 58, 940–951 (2026). https://doi.org/10.1038/s41588-026-02549-3
关键词: 全基因组精细定位, 致病基因变异, 复杂性状, 功能基因组注释, GWAS 方法