Clear Sky Science · zh
从第一性原理和机器学习分子动力学出发的酸性腐蚀微观动力学建模
为何金属在剧烈液体中更易生锈
从输油管道到汽车与船舶,许多关键结构由钢制成,而暴露于酸性水中时这些结构会悄然溶解。本文处理了一个长期存在的难题:如何从原子尺度向上预测铁在此类条件下的腐蚀速率,以及像锰这样的合金元素如何改变该速率。作者将量子计算与机器学习结合,构建了一个基于物理的详尽图景,描绘金属原子如何离开表面以及在此损失过程中氢气如何生成。
从原子到腐蚀速率的桥梁
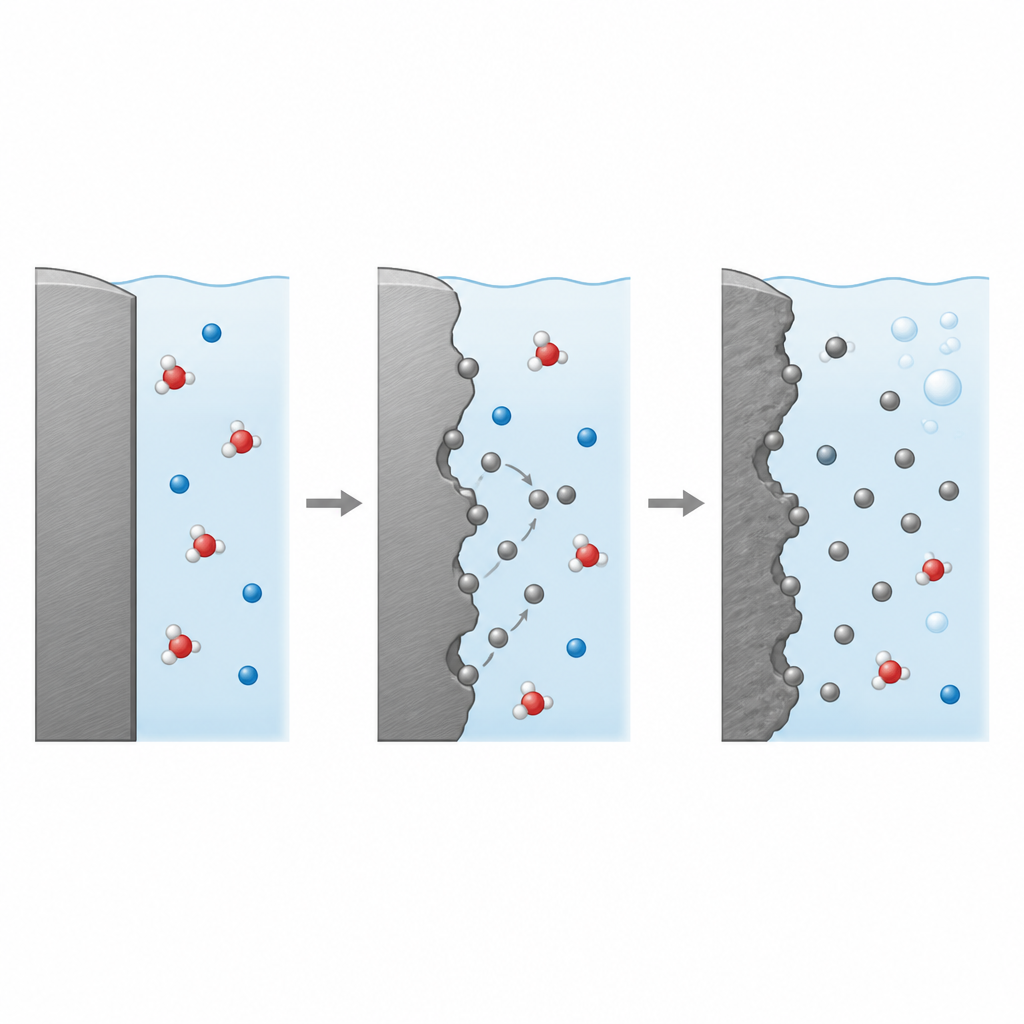
工程上长期使用经验公式来估算腐蚀,但这些方法常常掩盖真实的原子级过程。现有模型往往猜测能垒,忽略表面在不同电位和酸度下被水和反应物覆盖的情况,并将金属损失归结为单一步骤的多电子过程。与此不同,本研究构建了一个框架:从第一性原理的电子结构计算出发,随后使用机器学习驱动的分子动力学跟踪在真实金属—液体界面上运动的原子和水分子。这样研究组得以计算关键反应的能垒,并将这些能垒直接关联到可测量的电流和腐蚀速率。
铁原子如何逃离表面
作者首先解析了酸性溶液中铁原子从平坦铁表面脱离的过程。在这些条件下,水分子会吸附到表面、发生解离并形成短寿命的铁—氧—氢(FeOH)单元。研究表明,速度最慢、因此决定速率的步骤,是该吸附的FeOH单元失去电子并脱离进入溶液,随后成为被水分子包围的完全水合铁离子。通过使用增强采样技术和机器学习得到的相互作用模型跟踪自由能景观,研究者发现主控能垒约为0.76电子伏特。结合该能垒与精确计算的表面覆盖情况,他们的模型能够重现实验观测到的量,例如电流—电压曲线的斜率以及强酸中铁溶解的表观活化能。
从质子到气体:追踪氢气泡的形成
酸性腐蚀不仅关乎金属损失;它还产生氢气。因此,研究还分析了溶液中的质子在表面获取电子形成吸附氢原子并进一步结合成氢分子的步骤序列。采用相同的机器学习分子动力学方法,作者计算了三类经典步骤的能垒:初始的质子还原、一个质子与吸附氢反应的混合电化学步骤,以及两个吸附氢原子结合的纯化学步骤。他们的计算指向了这样一条路径:在相关电压窗口内,第一步——质子还原——是决定速率的步骤。有趣的是,模拟显示质子并非单纯从体相流体漫向表面,而是沿着水分子链以接力方式跳跃,呼应了液态水中著名的格罗特休斯(Grotthuss)机制。
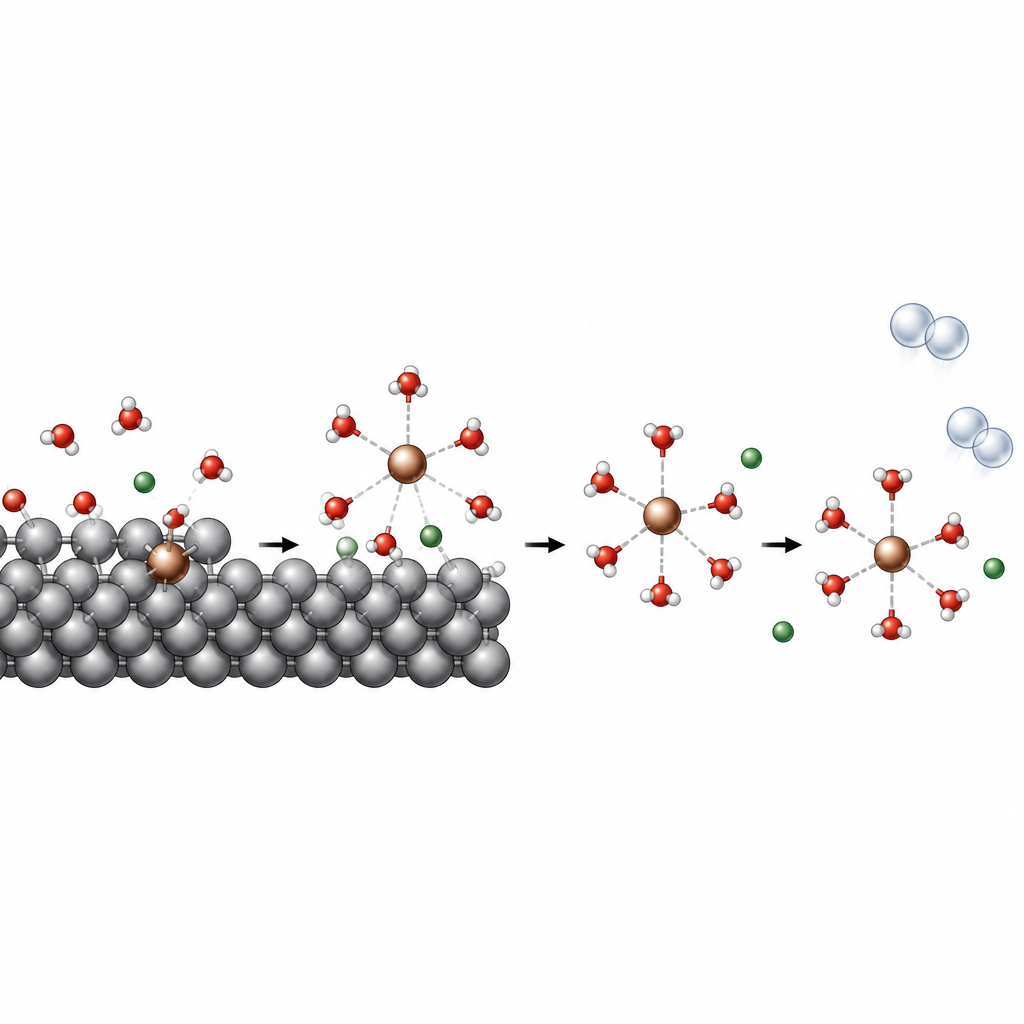
加入锰时发生了什么
钢通常含有锰以改善力学性能,但其对腐蚀的影响可能较为微妙。为探究这一点,作者在铁表面的外层引入一个锰原子并重复分析。在该锰位点附近,铁原子溶解的能垒和关键析氢步骤的能垒均被降低。当锰周围的局部行为按面积加权与周围铁的行为相结合时,整体腐蚀电流上升了几个数量级,腐蚀电位也向更负的方向移动。这些趋势与实验观察一致:富含锰的钢在酸性介质中往往腐蚀更快。
从详细模型到更安全的合金
通过证明原子尺度的能垒与现实的表面覆盖能够准确再现铁的测得腐蚀电流和电位,这项工作展示了一种强有力的方式来预测金属在酸中如何降解。原则上,相同的工作流程可以应用于其他金属、不同表面取向和合金元素以及不同的酸度水平。对非专业读者而言,关键讯息是:腐蚀不必被视为纯经验性的问题——借助现代计算与机器学习,可以在虚拟环境中测试合金成分与环境条件的设计选择如何影响关键基础设施的寿命。
引用: Bao, E., Xu, W., Ma, H. et al. Microkinetic modeling of acidic corrosion from first principles and machine-learning molecular dynamics. npj Comput Mater 12, 185 (2026). https://doi.org/10.1038/s41524-026-02047-4
关键词: 酸性腐蚀, 铁溶解, 析氢, 机器学习分子动力学, 合金设计