Clear Sky Science · zh
为通用机器学习原子间势优化跨域迁移
面向真实材料的更聪明模拟
设计新电池、催化剂和电子材料越来越依赖于追踪原子运动的计算模拟。基于量子力学的最可信模拟极其精确,但对于探索数百万个候选材料来说速度远远不够。更快的机器学习模型可以模拟量子计算,然而它们通常仅在狭窄情形下有效——例如只适用于晶体或只适用于分子。本文提出了一种构建单一通用模型(称为 SevenNet‑Omni)的方法,使其在金属表面、分子液体到多孔框架等多种材料类型中都保持高精度,同时仍足够快速以用于大规模发现。
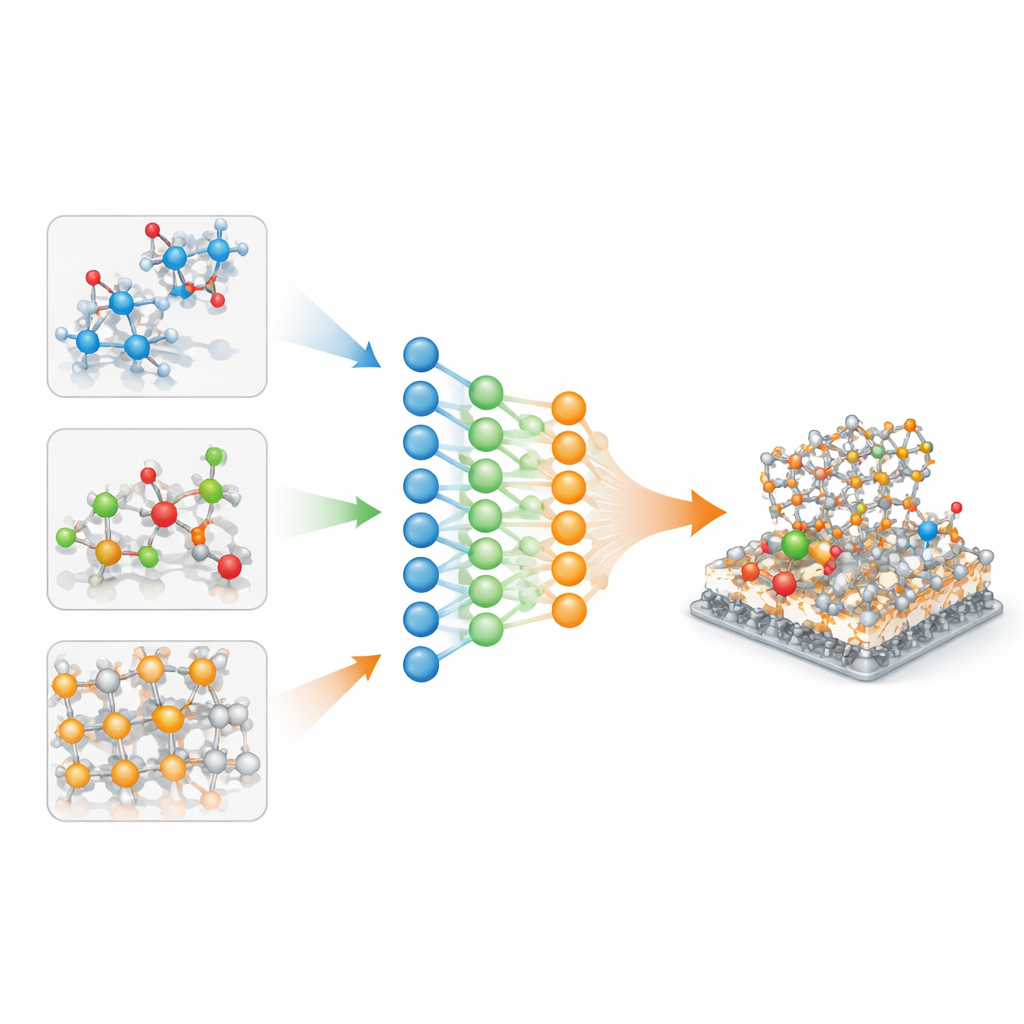
为何现有的原子模型难以迁移
当前的机器学习原子间势通常在单一精心整理的数据库上训练:一个用于无机晶体,另一个用于类药物分子,还有一个用于催化表面。每个数据库都采用各自的量子化学设定,因此底层的能量景观在细微的、非线性的方面有所不同。简单地把这些数据拼在一起——比如平移或缩放能量——会引入噪声,导致模型在其本域拟合良好,但在描述不熟悉的化学体系或稍有不同的量子方法时表现欠佳。随着材料问题越来越多地跨域混合,例如分子在固体表面溶液中反应,这种可迁移性差的问题已成为严重瓶颈。
共享骨干加温和的专用化
作者通过将每个数据库视为单一多任务神经网络中的一个“任务”来应对这一问题。在模型内部,一组参数构成共享骨干,捕捉原子键合的一般规律,而较小的任务特定参数则针对特定数据集微调行为。数学分析表明,如果任务特定部分过大,模型会有效地记忆每个数据库而忘记如何泛化。为防止这种情况,作者采用选择性正则化:直接惩罚大的任务特定参数,但允许共享骨干按需增长。这样推动网络尽可能通过共同物理原理来解释数据,仅对每个域做出适度修正。

用少量关键示例搭桥遥远领域
即便有正则化,某些化学空间区域只出现在单一数据库中,因此共享骨干在这些区域无法获得指导。为解决此问题,团队引入了“域桥集”。他们从若干数据库中精心挑选出极小的一部分配置——约千分之一——并用统一的量子力学设定重新计算这些配置。这些桥接结构就像语言教材中的双语短语:它们直接连接了两种不同量子方法对同一原子场景的描述。将它们纳入训练后,能强力拉紧任务之间的联系,使能量景观对齐,而无需对所有数据做暴力重算。系统性测试表明,正则化与桥接集相互增强,其联合效应超出任一方法单独能达到的改进。
构建并测试通用原子引擎
基于这些思想,作者在包含约2.42亿个原子结构的15个公共数据集上训练了 SevenNet‑Omni,覆盖了分子、晶体、催化剂、金属‑有机框架和多种量子理论层次。随后他们在熟悉与具有挑战性的情形中对模型进行了基准测试:晶体稳定性、金属中的晶界、钢材中的缺陷、类药物分子的扭转能垒、有机‑无机杂化钙钛矿、多孔框架中与碳捕集相关的吸附,以及金属表面的氢与二氧化碳转化反应。在这些测试中,SevenNet‑Omni 往往能匹配或超越为单一领域训练的专业模型,并在许多反应与吸附能量上保持“化学精度”。它还通过学习昂贵量子方法(r²SCAN)与更廉价、更丰富数据之间的关系,准确再现该昂贵方法的结果。
这对材料发现意味着什么
对非专家而言,核心信息是 SevenNet‑Omni 的表现更像一位在多个子领域都有经验的资深科学家。它不是过度拟合某一狭窄问题,而是学习广泛的化学原理并灵活地将其应用于新情形,从多孔固体中的气体捕集到金属电极上的反应。论文表明,这通过在数据集之间谨慎共享信息并轻度约束它们的差异,以及加入少量精心挑选的在量子方法之间“翻译”的示例是可行的。随着更大、更具多样性的数据集不断出现,这一训练策略为实现真正通用、可信赖的原子模型提供了一个可扩展的路径,能够加速化学、物理与材料科学领域的发现。
引用: Kim, J., You, J., Park, Y. et al. Optimizing cross-domain transfer for universal machine learning interatomic potentials. Nat Commun 17, 3432 (2026). https://doi.org/10.1038/s41467-026-70195-8
关键词: 机器学习原子间势, 多域材料建模, 迁移学习, 通用原子势, 材料发现