Clear Sky Science · fr
Optimisation du transfert inter‑domaines pour des potentiels interatomiques universels par apprentissage automatique
Des simulations plus intelligentes pour des matériaux réels
La conception de nouvelles batteries, de catalyseurs et de matériaux électroniques repose de plus en plus sur des simulations informatiques qui suivent le mouvement des atomes. Les simulations les plus fiables, basées sur la mécanique quantique, sont extrêmement précises mais beaucoup trop lentes pour explorer des millions de candidats. Des modèles d’apprentissage automatique plus rapides peuvent imiter les calculs quantiques, mais ils ne fonctionnent souvent que dans des situations restreintes — par exemple uniquement pour les cristaux ou uniquement pour les molécules. Cet article propose une manière de construire un modèle universel, appelé SevenNet‑Omni, qui reste précis pour de nombreux types de matériaux, des surfaces métalliques et liquides moléculaires aux structures poreuses, tout en restant suffisamment rapide pour la découverte à grande échelle.
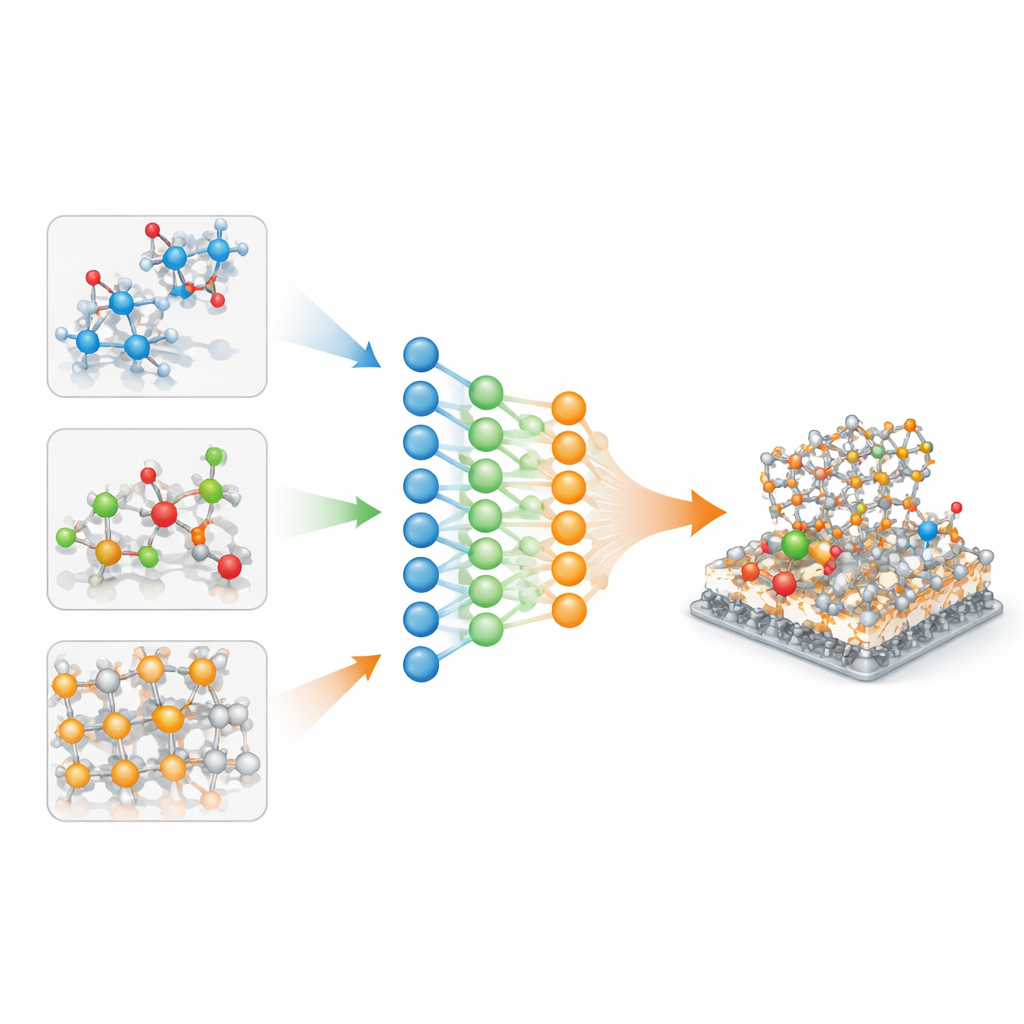
Pourquoi les modèles atomiques actuels ne voyagent pas bien
Les potentiels interatomiques par apprentissage automatique sont généralement entraînés sur une seule base de données soigneusement sélectionnée : une pour les cristaux inorganiques, une autre pour des molécules de type médicament, une autre encore pour les surfaces catalytiques. Chaque base est construite avec ses propres paramètres de chimie quantique, si bien que les paysages énergétiques sous‑jacents diffèrent de manière subtile et non linéaire. Simplement rassembler ces données — par exemple en décalant ou en redimensionnant les énergies — ajoute du bruit et conduit à des modèles qui s’ajustent bien à leur domaine d’origine mais échouent lorsqu’on leur demande de décrire des chimies inconnues ou des méthodes quantiques légèrement différentes. Alors que les problèmes en matériaux mêlent de plus en plus les domaines, comme des molécules réagissant sur des surfaces solides en solution, ce manque de transférabilité est devenu un goulot d’étranglement sérieux.
Une colonne vertébrale partagée avec une spécialisation douce
Les auteurs traitent chaque base de données comme une « tâche » distincte au sein d’un même réseau neuronal multi‑tâches. À l’intérieur du modèle, un ensemble de paramètres forme une colonne vertébrale partagée, capturant des règles générales de liaison atomique, tandis que de plus petits paramètres spécifiques à chaque tâche affinent le comportement pour des jeux de données particuliers. Une analyse mathématique montre que si les parties spécifiques à la tâche deviennent trop volumineuses, le modèle mémorise effectivement chaque base et oublie comment généraliser. Pour éviter cela, les auteurs appliquent une régularisation sélective : ils pénalisent directement les grands paramètres spécifiques aux tâches tout en laissant la colonne vertébrale partagée libre de croître si nécessaire. Cela incite le réseau à expliquer autant que possible par une physique commune, en n’utilisant que de modestes corrections pour chaque domaine.

Relier des mondes lointains avec quelques exemples clés
Même avec la régularisation, certaines régions de l’espace chimique n’apparaissent que dans une seule base, de sorte que la colonne vertébrale partagée ne reçoit aucune indication là‑dessus. Pour y remédier, l’équipe introduit un « ensemble de pontage inter‑domaines ». Ils sélectionnent avec soin une très faible fraction — environ un sur mille — de configurations issues de plusieurs bases de données et les recalculent avec un même protocole quantique. Ces structures de pontage agissent comme des phrases bilingues dans un manuel de langues : elles connectent directement la manière dont deux méthodes quantiques différentes décrivent une même scène atomique. Lorsqu’elles sont incluses à l’entraînement, elles renforcent fortement le lien entre les tâches, alignant les paysages énergétiques sans avoir à recalculer tout par force brute. Des tests systématiques montrent que la régularisation et l’ensemble de pontage se renforcent mutuellement, améliorant les performances au‑delà de ce que chacun peut accomplir seul.
Construire et tester un moteur atomique universel
Sur la base de ces idées, les auteurs entraînent SevenNet‑Omni sur 15 jeux de données publics comprenant environ 242 millions de structures atomiques, couvrant des molécules, des cristaux, des catalyseurs, des structures métal‑organiques poreuses et plusieurs niveaux de théorie quantique. Ils évaluent ensuite le modèle dans des situations familières et difficiles : stabilité des cristaux, joints de grains dans les métaux, défauts dans les aciers, barrières de torsion dans des molécules de type médicament, pérovskites hybrides organiques‑inorganiques, adsorption dans des structures poreuses pertinentes pour la capture du carbone, et réactions sur surfaces métalliques importantes pour la conversion de l’hydrogène et du dioxyde de carbone. Dans ces tests, SevenNet‑Omni rivalise souvent avec, voire dépasse, des modèles spécialisés entraînés pour un seul domaine, et il maintient une « précision chimique » pour de nombreuses énergies de réaction et d’adsorption. Il reproduit également fidèlement des résultats d’une méthode quantique coûteuse (r²SCAN) en apprenant comment cette méthode se rapporte à des données moins onéreuses et plus abondantes.
Ce que cela signifie pour la découverte de nouveaux matériaux
Pour les non‑spécialistes, le message principal est que SevenNet‑Omni se comporte comme un chercheur expérimenté ayant travaillé dans de nombreux sous‑domaines. Plutôt que de surajuster un problème étroit, il apprend des principes chimiques généraux et les applique de manière flexible à de nouvelles situations, de la capture de gaz dans des solides poreux aux réactions sur électrodes métalliques. L’article montre que cela est possible en partageant soigneusement l’information entre jeux de données tout en contraignant légèrement leurs différences, et en ajoutant un petit nombre d’exemples de « traduction » choisis entre méthodes quantiques. À mesure que des bases plus larges et plus diverses apparaîtront, cette stratégie d’entraînement offre une voie évolutive vers des modèles atomiques véritablement universels et fiables, capables d’accélérer la découverte en chimie, physique et science des matériaux.
Citation: Kim, J., You, J., Park, Y. et al. Optimizing cross-domain transfer for universal machine learning interatomic potentials. Nat Commun 17, 3432 (2026). https://doi.org/10.1038/s41467-026-70195-8
Mots-clés: potentiels interatomiques par apprentissage automatique, modélisation multi‑domaine des matériaux, apprentissage par transfert, potentiel atomistique universel, découverte de matériaux